 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Katarina Trebušak Podkrajšek | + 2963 word(s) | 2963 | 2020-09-22 05:12:48 | | | |
| 2 | Tina Levstek | Meta information modification | 2963 | 2020-09-27 20:05:40 | | |
Video Upload Options
Fabry disease (FD; OMIM#301500) is an X-linked lysosomal storage disorder associated with inherited or de novo disease causing variants in the α-galactosidase A gene (GLA; OMIM*300644). Reduced or even absent α-galactosidase A (α-Gal A; EC 3.2.1.22) activity leads to accumulation of glycosphingolipids with terminal α-D-galactosyl residues, especially globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) in plasma, urine and different organ systems, mainly cardiac, renal, endothelial and neuronal. The major physiological source of Gb3 is globoside, a glycolipid of erythrocytes and cells membranes found in different tissues.
Kidneys are very frequently affected in patients with Fabry disease regardless of gender. Most important manifestations of Fabry nephropathy are proteinuria and slowly progressive chronic kidney disease, which can in some cases lead to end stage renal disease.
1. Introduction
FD is rare disorder with the estimated incidence in the general population between 1 in 40,000 and 1 in 117,000.[1] However, based on recent newborn screening studies, the prevalence in some populations was reported to be markedly higher with 1 in 1300 to 1 in 7800 males.[2]
Pathophysiological processes start at a very young age,[3] but most patients do not show symptoms in infancy. Gb3 deposition has been detected in fetuses[4] and in newborns with classical FD phenotype.[3] Men with classical phenotype, who have no or very low α-Gal A activity (<1% of mean normal), often develop first symptoms in early childhood, including acroparesthesias, angiokeratomas, intolerance to heat, hypo/anhidrosis, cornea verticillate and gastrointestinal disturbances.[5][6][7] The disease then gradually progresses to major organ failure in early adulthood, such as progressive kidney disease, cardiac signs, and symptoms and cerebrovascular events.[5][6][7] On the other hand, men who have a significant residual α-Gal A activity typically present with only one organ involvement.[6],[7] Although, FD is an X-linked trait, women can also develop typical symptoms due to random X-inactivation, which result in a mosaicism of gene expression, leading to differential expression of the functional or mutant enzyme.[8] Inactivation of the mutant allele leads to a milder phenotype, while the inactivation of the wild-type allele leads to more severe phenotype with an earlier onset.[9] The unaffected cells secrete mostly 46 kDa mature form of the α-Gal A and not the high-uptake mannose 6-phosphorilated form. The mature form is able to complement the activity in the population of cells lacking the expression of the enzyme.[10] Clinical presentation in women therefore varies from an asymptomatic or mild, later onset phenotype to a phenotype similar to that of men with a classical phenotype.[11]
The diagnosis of FD in men is based on reduced α-Gal A activity in plasma, leukocytes, or dried blood spots,[12][13][14] but in women, this measurement is unreliable, because enzyme activity in female with FD may not be elevated.[15] Gb3, the substrate of α-Gal A and the main accumulation deposit in FD, was considered as a possible diagnostic marker of FD. Gb3 accumulates mainly within cells, while it circulates in the extracellular space within lipoproteins.[16] However, there is no strict correlation between plasma Gb3 concentrations and clinical manifestation in Fabry patients.[17][18] Therefore, Gb3 level in urine and plasma are only suitable for identifying males with classical phenotype. Elevated plasma lyso-Gb3, the deacylated derivative of Gb3, has been designated as a hallmark of FD[19] and allows better differentiation between patients with classical and non-classical phenotype and healthy subjects.[20][21][22] Therefore, this biomarker may improve the diagnosis of clinically relevant FD, particularly in females with normal or borderline α-Gal A activity.[21] However, false negative results were also reported in non-classical phenotypes.[19][21] After the introduction of enzyme replacement therapy (ERT), patients with classical phenotypes show a rapid decrease in lyso-Gb3 concentration, whereas the decrease is slower in men with non-classical phenotype and women.[23][24] The serum lyso-Gb3 may serve as a marker for tissue involvement to assess which heterozygotes should be considered for treatment despite normal α-Gal A activity.[21] The gold standard for the diagnosis of female Fabry patients is still GLA gene sequencing, which is also important in men for the confirmation of the pathogenic GLA variant and prediction of the disease phenotype [28].[25] Early diagnosis of FD is important to initiate Fabry disease specific treatment, namely with ERT[24] or chaperones.[26]
2. Current Diagnostic Approaches and Follow-Up of Fabry Nephropathy
Before the era of kidney transplantation and dialysis, end-stage renal disease (ESRD) was the leading cause of premature death in patients with FD.[1] However, progressive nephropathy remains one of the main manifestations of FD, which usually leads to ESRD in untreated patients with classical phenotype from the third to the fifth decade of life.[27][28] The rate of progression of chronic kidney disease (CKD) is similar to that of diabetic nephropathy.[29] The Fabry Outcome Survey reported a baseline prevalence of nephropathy in 59% of men and 38% of women with FD.[1] Even though progression to ESRD is less common in women with FD and are less likely to progress from moderate CKD to ESRD, the median age at which patients reached ESRD was 38 years regardless of their gender.[11][27] Renal involvement contributes largely to the overall burden of morbidity and mortality in FD.[6]
Gb3 accumulation occurs in various types of renal cells, including podocytes, mesangial, and interstitial cells, cells of the proximal and distal tubules, and loop of Henle, as well as vascular endothelial cells and smooth muscle cells.[30] The understanding of the pathophysiological mechanisms leading to CKD and ESRD is still scarce and further research is needed to clarify the complexity of genotype-phenotype correlation.
The main approaches used in nephrology to diagnose patients with FD are kidney biopsy, high-risk population testing, and family screening.[31] FD should be considered in patients with CKD without a clear cause of nephropathy.[2] For diagnosis, assessment of renal involvement, and monitoring of treatment, biomarkers of renal damage are often evaluated (albuminuria/proteinuria, serum creatinine, glomerular filtration rate (GFR), and cystatin C) together with urinary microscopy and renal biopsy.
2.1. Albuminuria/Proteinuria
Albuminuria/proteinuria is clinically most often used biomarker of Fabry nephropathy, although kidney damage is present already in the non-albuminuric state. Therefore they are not sensitive biomarkers for early kidney damage, as biopsies of normoalbuminuric Fabry patients have already shown advanced lesions.[32] However, proteinuria is one of the most important markers for monitoring the progression of nephropathy in treated and untreated patients, since patients with higher proteinuria show a much steeper decline in renal function over time.[27][29] Proteinuria has been shown to stimulate interstitial inflammation and fibrosis. Furthermore it promotes tubular cells to undergo partial epithelial mesenchymal trans-differentiation, which induces cell-cycle arrest and promotes the release of fibrogenic cytokines.[33][34]
2.2. Serum Creatinine and Glomerular Filtration Rate (GFR)
Regular assessment of renal function in Fabry patients includes use of measured and estimated glomerular filtration rate (eGFR). Due to inaccuracy of creatinine-based GFR measurements, it is recommended to use measured GFR measurements (e.g., iohexol GFR) at least annually.[34] Because these methods are more complex and tedious, estimated GFR measurements using appropriate formula are more widely used. Currently used serum creatinine-based equations in the clinical practice are the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation for adults[35] and the Schwartz formula for children.[36] Some Fabry patients develop GFR loss before development of proteinuria.[11] Glomerular hyperfiltration may be a common feature in young Fabry patients and may be considered as an early marker of Fabry nephropathy,[37] which masks impairment of renal function, although rare, a decline in GFR can already be seen in adolescence.[38] Slope of progression of renal insufficiency was correlated to the level of proteinuria and in addition, it was not linear as shown by Schiffmann et al., where they retrospectively analyzed the decline of eGFR. The slope of renal function with an eGFR of more than 60 mL/min/1.73 m2 was −3.0 in men and −0.9 mL/min/1.73 m2/year in women, and with an eGFR of less than 60 mL/min/1.73 m2 it was −6.8 and −2.1 mL/min/1.73 m2/year in men and women, respectively.[39]
2.3. Cystatin-C
Cystatin-C, a cysteine protease inhibitor, is constantly produced by all nucleated cells. It is freely filtered by the glomerulus and then reabsorbed and catabolized in the tubular epithelial cells. Therefore, it does not re-enter the bloodstream or urine. Cystatin-C concentration was found as a superior and more sensitive marker than serum creatinine for detecting early renal dysfunction and small decreases in GFR in Fabry patients of both genders.[40] Therefore, it could be valuable as a prognostic marker and for estimating the efficiency of the ERT.[40] Probably the main reason why cystatin-C is not widely used is that it is more costly, time-consuming, and less available than creatinine.
2.4. Urine Microscopy
As a non-invasive, reasonably priced, and expeditious diagnostic tool, urine microscopy can be useful for the diagnosis and assessment of FD progression. Most of the cells present in the urine of Fabry patients are renal tubular epithelial cells.[41] Mulberry cells with characteristic “Maltese cross bodies” (oval fat bodies) can be detected in the urinary sediments of Fabry patients under a polarized microscope.[42][43] Example of a Maltese cross in the urine sediment of FD patients is shown in Figure 1. Furthermore, there is a specific morphological population of Maltese cross bodies, characterized by a lamellarized appearance with protrusions, probably due to Gb3 resembling “mosquito coils”, which most likely represents a fragmentation of shed nephronal epithelial cells with accumulated lysosomal Gb3.[44] In addition their excretion correlated with the concentration of albumin in urine and could therefore be useful for accessing Fabry nephropathy burden.[44] Despite the fact, that this method could represent a valuable tool it is not commonly used, because it requires special equipment (phase contrast microscope) and well-trained personnel.

Figure 1. Birefringent Maltese cross in the urine sediment of Fabry patient when viewed under a polarized microscope (magnification 400×). Figure courtesy of Mravljak M; Department of Internal Medicine, General Hospital Slovenj Gradec.
2.5. Renal Biopsy
Kidney biopsy with electron microscope analysis is recommended for all individuals with CKD, a GLA variant of unknown significance, and an uncertain diagnosis of FD to rule out other comorbidities, as it is currently the only reliable diagnostic method for confirming or excluding Fabry nephropathy.[45] Significant histologic changes occur before the typical clinical signs of CKD therefore, findings in biopsy are crucial for choosing the optimal therapeutic strategy and follow-up in high-risk patients.[46][47][48][49] Example of a kidney biopsy under electron microscopy with typical lamellar inclusions in a 27 year old Fabry male patient with normal kidney function (eGFR 102 mL/min/1.73 m2), normoalbuminuria (albumin to creatinine ratio (ACR) 12 mg/g), but high levels of podocyturia (urinary podocytes (uPod) 2420/g creatinine) is shown in Figure 2. Electron microscopic studies demonstrate typical osmophilic bodies, called myeloid or Zebra bodies, packed with lamellated membrane structures.[30] In contrast to electron microscopy, diagnosis could be much more challenging with light microscopy techniques, since the majority of stainings cause a washout of lipid contents. As a result, more unspecific vacuolization of podocytes and epithelial cells is a characteristic histological finding.[30][50] Mesangial expansion, segmental and global glomerulosclerosis, tubular atrophy, and intestinal fibrosis are also present at an early stage.[30][50][51] To better classify the extent of tissue involvement, the International Study Group of Fabry Nephropathy developed a scoring system of histological changes on light microscopy and toluidine blue–stained semithin sections.[49] The score quantifies the Gb3 density deposition in glomeruli, interstitium, and vessels, as well as progressive lesions (glomerulosclerosis, ischemic glomeruli, and tubulointerstitial fibrosis). Recently, bedside stereomicroscopy for Fabry kidney biopsies has been recommended as a complementary method to current histologic evaluation as it was demonstrated to have high diagnostic sensitivity for FD.[52]
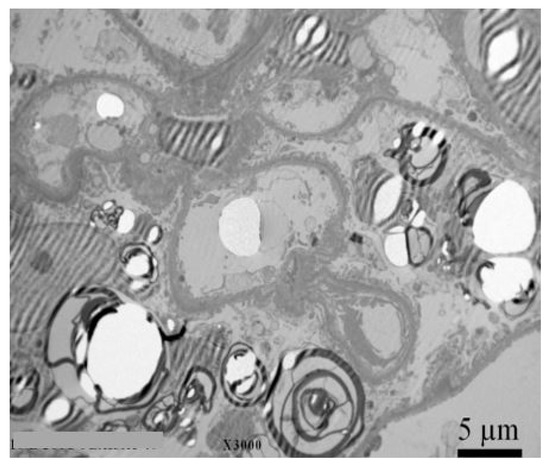
Figure 2. Kidney biopsy with electron microscopy with typical lamelar inclusions in a 27-year old Fabry male patient with normal kidney function (eGFR 102 mL/min/1.73 m2), normoalbuminuria (ACR 12 mg/g), but high levels of podocyturia (UPod 2.420/g creatinine). Diffuse and numerous myeloid inclusions in podocyte cytoplasm (black arrow) with presence of vacuoles in the cytoplasm of podocytes (white arrow) are evident. Figure courtesy of Pleško J and Kojc N; Institute of Pathology, Ljubljana Medical Faculty.
3. Novel Biomarkers for Predicting Development and Progression of Fabry Nephropathy
Heterogeneous phenotype, varying disease severity and symptoms onset make the diagnosis of FD notoriously difficult and often delayed for several years. Early diagnosis is crucial, as treatment should begin before the kidneys are irreversibly damaged. ERT was reported to reduce Gb3 deposits from most renal cells, but to a lesser extent in podocytes,[53] and to stop or slow down the progression of Fabry nephropathy.[54][55][56] However, once proteinuria occurs, it usually does not normalize despite treatment.[57] In addition, in patients with overt proteinuria or reduced GFR (<60 mL/min/1.73 m2), ERT does not prevent further deterioration of renal function.[2] Therefore, there is a considerable need for novel biomarkers that would enable identification and also prediction of development and progression of Fabry nephropathy.
Patients with rapid progression of kidney disease have higher urinary protein to creatinine ratio[58] and additionally, underlying conditions, such as hypertension, hyperlipidemia, and smoking, may contribute to a progressive loss of GFR.[59] However, environmental factors and glycolipid accumulation due to the disease causing GLA variants cannot fully explain the phenotypic variability. Even between the same family members carrying the same pathogenic GLA variant, there is enormous clinical variability.[60] It is therefore reasonable to anticipate that additional unknown biochemical, genetic, and epigenetic factors (modifiers) may influence the rate of progression of Fabry nephropathy. Recently, new biomarkers (bikunin, tubular proteins) have been proposed to improve the assessment of renal impairment, but further research is needed to evaluate their clinical utility.
3.1. Bikunin
Bikunin or urinary trypsin inhibitor is a serine protease inhibitor present in plasma and many tissues. It is excreted in urine and its levels were found significantly higher in Fabry patients with nephropathy; therefore, it may serve as a biomarker of renal impairment in FD.[61] The origin of the higher bikunin levels may imply direct renal involvement and secondary activation in response to the storage of glycosphingolipids in biochemical pathways associated with inflammation; however, further studies are needed to elucidate the mechanisms involved in the elevation of urinary bikunin levels.[61] Yet, renal impairment alone is not sufficient to explain higher urinary bikunin levels, as no correlation between serum creatinine and urinary bikunin levels was found.[61]
3.2. Tubular and Glomerular Proteins
Impaired glomerular and tubular function in Fabry patients is reflected in an abnormal urinary excretion of tubular and glomerular proteins. Therefore, these biomarkers could be a valuable tool to assess kidney involvement and predict the progression of Fabry nephropathy. Larger studies are needed to thoroughly investigate the sensitivity and their correlations with Fabry nephropathy progression, the biomarkers currently used, and the changes in response to ERT.
Aguiar et al. found that the biomarkers of glomerular (transferrin and type IV collagen) and tubular (α1-microglobulin, N-acetyl-β-glucosaminidase, and alanine aminopeptidase) dysfunction were elevated even in a subgroup of patients without clinical signs of kidney disease. Furthermore, more significant correlation with eGFR was reported for type IV collagen and N-acetyl-β-glucosaminidase as it was for albuminuria.[62] Besides increased N-acetly-β-D-glucosaminidase, Shiffmann et al. also reported an increase in β2-microglobulin.[63] A decrease in the glomerular marker IgG, the tubular markers α1-microglobulin and retinol-binding protein as well as the shared tubular and glomerular markers albumin and transferrin in a population of 13 women with FD after long-term ERT was reported.[64] Furthermore another study showed normalization of urinary excretion of uromodulin after ERT and reduction in untreated Fabry patients.[65]
3.3. Urine Podocytes
The earliest sign of renal damage appears to be podocyte foot process effacement, whereas it was found in young classic FD patients without clinically evident signs of Fabry nephropathy.[32][66][67] Podocytes as terminally differentiated cells do not divide; therefore, their replacement potential in adult is limited. Podocytes accumulate Gb3 more than other renal cell types.[30] Accumulation in podocytes continues until the third decade.[68] Alongside their volume continues to increase, resulting in an increase in podocyte foot process width and podocyte loss.[68] Injured podocytes detach from the glomerular basement membrane and are lost to urine. Podocyturia therefore correlates with the clinical severity of Fabry nephropathy.[67] A recent study of podocyte glycocalyx damage showed that podocalyxin loss may be associated with reduced adhesion of podocytes to the extracellular matrix, which enables detachment and urinary excretion.[69] Podocyturia disrupts glomerular permselectivity, causes albuminuria/proteinuria and leads to glomerulosclerosis and fibrosis.[70][71] A positive correlation was found between podocyturia and ACR.[72] Despite podocyturia is an early clinical sign of kidney injury and could serve as a diagnostic test to assess kidney involvement, it is still not regularly used in clinical practice. This is mainly because methods for assessing podocyturia are not yet standardized and not available in the majority of clinical settings.
4. Future Perspective
Considering recent advances in sequencing approaches, it is reasonable to anticipate, that novel genomic and transcriptomics markers influencing the development and progression of the nephropathy are going to be identified. The metabolic origin and rate of progression of Fabry nephropathy resembles that of diabetic nephropathy. Since several studies have already been conducted in the field of CKD and diabetic nephropathy,[73] translating of the knowledge of biomarkers suspected of being involved in the development and progression of kidney failure could contribute to a better understanding of the molecular mechanisms of Fabry nephropathy. The development of new -omics technologies has led to new opportunities in the search for novel biomarkers. Despite several promising biomarkers, so far none of them have been translated into clinical practice. It is highly unlikely that the biomarkers discovered with -omics technologies will alone be sufficient to reliably predict the development and/or progression of Fabry nephropathy. Multifactorial models that would integrate clinical, genetic, and biochemical factors[74] are more likely to provide the evidence needed to translate this knowledge into clinical practice.
Abbreviations
- α-Gal A - α-galactosidase A
- GLA - α-galactosidase A gene
- ACR - albumin to creatinine ratio
- CKD - chronic kidney disease
- eGFR - estimated glomerular filtration rate
- ERT - enzyme replacement therapy
- ESRD - end stage renal disease
- FD - Fabry disease
- Ga2 - galabiosylceramide
- Gb3 - globotriaosylceramide
- lyso-Gb3 - globotriaosylsphingosine
- UPod - urinary podocytes
References
- Atul Mehta; J T R Clarke; R Giugliani; P Elliott; Aleš Linhart; M Beck; Gere Sunder-Plassmann; on behalf of the FOS Investigators; Natural course of Fabry disease: changing pattern of causes of death in FOS - Fabry Outcome Survey. Journal of Medical Genetics 2009, 46, 548-552, 10.1136/jmg.2008.065904.
- Raphael Schiffmann; Derralynn Hughes; Gabor E. Linthorst; Alberto Ortiz; Einar Svarstad; David G. Warnock; Michael L. West; Christoph Wanner; Daniel-G Bichet; Erik Ilsø Christensen; et al.Ricardo Correa-RotterP M ElliottSandro FeriozziAgnes B. FogoDominique P. GermainCarla E.M. HollakRobert J. HopkinJack JohnsonIlkka KantolaJeffrey B. KoppJürgen KrönerAleš LinhartAna Maria MartinsDietrich MaternAtul C. MehtaRenzo MignaniBehzad NajafianIchiei NaritaKathleen NichollsGreg T. ObradorJoão Paulo OliveiraAntonio PisaniJuan M. PoliteiUma RamaswamiMarkus RiesW. TerrynCamilla TøndelRoser TorraBojan VujkovacS. WaldekJerry Walter Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney International 2017, 91, 284-293, 10.1016/j.kint.2016.10.004.
- A. C. Vedder; A. Strijland; M. A. Vd Bergh Weerman; S. Florquin; Johannes M. F. G. Aerts; C. E. M. Hollak; Manifestations of Fabry disease in placental tissue. Journal of Inherited Metabolic Disease 2006, 29, 106-111, 10.1007/s10545-006-0196-0.
- Subhash Popli; David J. Leehey; Zelma V. Molnar; Zeenat M. Nawab; Todd S. Ing; Demonstration of Fabry's disease deposits in placenta. American Journal of Obstetrics and Gynecology 1990, 162, 464-465, 10.1016/0002-9378(90)90410-9.
- João Paulo Oliveira; Susana Ferreira; Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype- phenotype correlations.. The Application of Clinical Genetics 2019, 12, 35-50, 10.2147/TACG.S146022.
- Dominique P. Germain; Fabry disease. Orphanet Journal of Rare Diseases 2010, 5, 30-30, 10.1186/1750-1172-5-30.
- Robert J. Desnick; Fabry disease: α-galactosidase A deficiency. Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease 2020, 2015, 575-587, 10.1016/b978-0-12-813955-4.00042-8.
- I Redonnet-Vernhet; J K Ploos Van Amstel; R P Jansen; Ron Wevers; R Salvayre; T Levade; Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene.. Journal of Medical Genetics 1996, 33, 682-688, 10.1136/jmg.33.8.682.
- Robert Dobrovolny; Lenka Dvorakova; Jana Ledvinova; SudheerA Magage; Jan Bultas; Jean-Claude M. Lubanda; Milan Elleder; Debora Karetová; Marketa Pavlikova; M. Hrebícek; et al. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the α-galactosidase A gene in the Czech and Slovak population. Journal of Molecular Medicine 2005, 83, 647-654, 10.1007/s00109-005-0656-2.
- Michael Beck; Timothy M. Cox; Comment: Why are females with Fabry disease affected?. Molecular Genetics and Metabolism Reports 2019, 21, 100529, 10.1016/j.ymgmr.2019.100529.
- William R. Wilcox; João Paulo Oliveira; Robert J. Hopkin; Alberto Ortiz; Maryam Banikazemi; Ulla Feldt-Rasmussen; Katherine Sims; Stephen Waldek; Gregory M. Pastores; Philip Lee; et al.Christine M. EngLászló MaródiKevin E. StanfordFrank BreunigChristoph WannerDavid G. WarnockRoberta LemayDominique P. GermainAlberto Ortiz Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Molecular Genetics and Metabolism 2008, 93, 112-128, 10.1016/j.ymgme.2007.09.013.
- Andreas Gal; D Hughes; Bryan G. Winchester; Toward a consensus in the laboratory diagnostics of Fabry disease - recommendations of a European expert group. Journal of Inherited Metabolic Disease 2011, 34, 509-514, 10.1007/s10545-010-9261-9.
- Luca Massaccesi; Alberto Burlina; Claudia J. Baquero; G. Goi; Alessandro P Burlina; Guido Tettamanti; Whole-blood alpha-D-galactosidase A activity for the identification of Fabry's patients. Clinical Biochemistry 2011, 44, 916-921, 10.1016/j.clinbiochem.2011.03.141.
- Markus A. Hölzl; Miriam Gärtner; Johannes J. Kovarik; Johannes Hofer; Hanno Bernheimer; Gere Sunder-Plassmann; Gerhard J. Zlabinger; Quantification of α-galactosidase activity in intact leukocytes. Clinica Chimica Acta 2010, 411, 1666-1670, 10.1016/j.cca.2010.06.023.
- Gabor E. Linthorst; Anouk C. Vedder; Johannes M. F. G. Aerts; Carla E.M. Hollak; Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clinica Chimica Acta 2005, 353, 201-203, 10.1016/j.cccn.2004.10.019.
- Hernán Trimarchi; R. Canzonieri; A. Schiel; C. Costales-Collaguazo; Juan M. Politei; A. Stern; M. Paulero; T. Rengel; J. Andrews; M. Forrester; et al.M. LombiV. PomeranzR. IriarteA. MuryanElsa ZottaMaria Dolores Sanchez-NiñoAlberto Ortiz Increased urinary CD80 excretion and podocyturia in Fabry disease. Journal of Translational Medicine 2016, 14, 289, 10.1186/s12967-016-1049-8.
- A. C. Vedder; G. E. Linthorst; M. J. Van Breemen; J. E. M. Groener; F. J. Bemelman; A. Strijland; M. M. A. M. Mannens; Johannes M. F. G. Aerts; Carla Hollak; The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels. Journal of Inherited Metabolic Disease 2007, 30, 68-78, 10.1007/s10545-006-0484-8.
- Soumeya Bekri; Olivier Lidove; Roland Jaussaud; Bertrand Knebelmann; Fréderic Barbey; The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry disease: a review of the literature.. Cardiovascular & Hematological Agents in Medicinal Chemistry 2006, 4, 289-297, 10.2174/187152506778520718.
- Johannes M. F. G. Aerts; Johanna E. Groener; Sijmen Kuiper; Wilma E. Donker-Koopman; Anneke Strijland; Roelof Ottenhoff; Cindy Van Roomen; Mina Mirzaian; Frits A. Wijburg; Gabor E. Linthorst; et al.Anouk C. VedderSaskia M. RombachJosanne Cox-BrinkmanPentti SomerharjuRolf G. BootCarla E. HollakRoscoe O. BradyBen J. Poorthuis Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proceedings of the National Academy of Sciences 2008, 105, 2812-2817, 10.1073/pnas.0712309105.
- S.M. Rombach; N. Dekker; M.G. Bouwman; G.E. Linthorst; A.H. Zwinderman; F.A. Wijburg; S. Kuiper; M.A. Vd Bergh Weerman; J.E.M. Groener; B.J. Poorthuis; et al.C.E.M. HollakJohannes M. F. G. Aerts Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2010, 1802, 741-748, 10.1016/j.bbadis.2010.05.003.
- Albina Nowak; Thomas P. Mechtler; R.J. Desnick; David Kasper; Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Molecular Genetics and Metabolism 2017, 120, 57-61, 10.1016/j.ymgme.2016.10.006.
- Tadayasu Togawa; Takashi Kodama; Toshihiro Suzuki; Kanako Sugawara; Takahiro Tsukimura; Toya Ohashi; Nobuyuki Ishige; Ken Suzuki; Teruo Kitagawa; Hitoshi Sakuraba; et al. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Molecular Genetics and Metabolism 2010, 100, 257-261, 10.1016/j.ymgme.2010.03.020.
- Hitoshi Sakuraba; Tadayasu Togawa; Takahiro Tsukimura; Hiroshi Kato; Plasma lyso-Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clinical and Experimental Nephrology 2017, 22, 843-849, 10.1007/s10157-017-1525-3.
- Maarten Arends; Marieke Biegstraaten; Christoph Wanner; Sandra Sirrs; Atul Mehta; Perry M Elliott; Daniel Oder; Oliver T Watkinson; Daniel G Bichet; Aneal Khan; et al.Mark IwanochkoFrédéric M VazAndré B P Van KuilenburgMichael L WestDerralynn A HughesCarla E M Hollak Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. Journal of Medical Genetics 2018, 55, 351-358, 10.1136/jmedgenet-2017-104863.
- Alberto Ortiz; Dominique P. Germain; Robert J. Desnick; Juan Politei; Michael Mauer; Alessandro Burlina; Christine Eng; Robert J. Hopkin; Dawn Laney; Aleš Linhart; et al.Stephen WaldekEric WallaceFrank WeidemannWilliam R. Wilcox Fabry disease revisited: Management and treatment recommendations for adult patients. Molecular Genetics and Metabolism 2018, 123, 416-427, 10.1016/j.ymgme.2018.02.014.
- Jian-Qiang Fan; Satoshi Ishii; Active-site-specific chaperone therapy for Fabry disease. The FEBS Journal 2007, 274, 4962-4971, 10.1111/j.1742-4658.2007.06041.x.
- Alberto Ortiz; João P Oliveira; Steven Waldek; David G Warnock; Bruno Cianciaruso; Christoph Wanner; On Behalf Of The Fabry Registry; Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrology Dialysis Transplantation 2008, 23, 1600-1607, 10.1093/ndt/gfm848.
- A. Pisani; B. Visciano; M. Imbriaco; A. Di Nuzzi; A. Mancini; C. Marchetiello; E. Riccio; The kidney in Fabry's disease. Clinical Genetics 2014, 86, 301-309, 10.1111/cge.12386.
- Alberto Ortiz; João Paulo Oliveira; Christoph Wanner; Barry M Brenner; Stephen Waldek; David G Warnock; Alberto Ortiz; Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nature Clinical Practice Nephrology 2008, 4, 327-336, 10.1038/ncpneph0806.
- Joseph Alroy; Sharda Sabnis; Jeffrey B. Kopp; Renal Pathology in Fabry Disease. Journal of the American Society of Nephrology 2002, 13, S134-S138, 10.1097/01.asn.0000016684.07368.75.
- Bojan Vujkovac; Fabry disease: diagnostic methods in nephrology practice. Clinical Nephrology 2017, 88, 44-47, 10.5414/cnp88fx28.
- Camilla Tøndel; Leif Bostad; Asle Hirth; Einar Svarstad; Renal Biopsy Findings in Children and Adolescents With Fabry Disease and Minimal Albuminuria. American Journal of Kidney Diseases 2008, 51, 767-776, 10.1053/j.ajkd.2007.12.032.
- Xiao-Ming Meng; David J. Nikolic-Paterson; Hui-Yao Lan; Inflammatory processes in renal fibrosis. Nature Reviews Nephrology 2014, 10, 493-503, 10.1038/nrneph.2014.114.
- Dong Zhou; Youhua Liu; Understanding the mechanisms of kidney fibrosis. Nature Reviews Nephrology 2015, 12, 68-70, 10.1038/nrneph.2015.215.
- Andrew S Levey; Lesley A. Stevens; Christopher H. Schmid; Yaping (Lucy) Zhang; Alejandro F. Castro; Harold I. Feldman; John W. Kusek; Paul Eggers; Frederick Van Lente; Tom Greene; et al.Josef Coreshfor the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A New Equation to Estimate Glomerular Filtration Rate. Annals of Internal Medicine 2009, 150, 604-612, 10.7326/0003-4819-150-9-200905050-00006.
- George J. Schwartz; Alvaro Muñoz; Michael F. Schneider; Robert H. Mak; Frederick Kaskel; Bradley A. Warady; Susan L. Furth; New Equations to Estimate GFR in Children with CKD. Journal of the American Society of Nephrology 2009, 20, 629-637, 10.1681/asn.2008030287.
- Eleonora Riccio; Massimo Sabbatini; Dario Bruzzese; Luigi Annicchiarico Petruzzelli; Angela Pellegrino; Letizia Spinelli; Roberta Esposito; Massimo Imbriaco; Sandro Feriozzi; Antonio Pisani; et al.on behalf of AFFIINITY Group Glomerular Hyperfiltration: An Early Marker of Nephropathy in Fabry Disease. Nephron 2018, 141, 10-17, 10.1159/000493469.
- Uma Ramaswami; Behzad Najafian; Arrigo Schieppati; Michael Mauer; Daniel Bichet; Assessment of renal pathology and dysfunction in children with Fabry disease.. Clinical Journal of the American Society of Nephrology 2010, 5, 365-70, 10.2215/CJN.08091109.
- Raphael Schiffmann; David G. Warnock; Maryam Banikazemi; Jan Bultas; Gabor E. Linthorst; Seymour Packman; Sven Asger Sorensen; William R. Wilcox; R.J. Desnick; Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrology Dialysis Transplantation 2009, 24, 2102-2111, 10.1093/ndt/gfp031.
- Miguel-Ángel Torralba-Cabeza; Susana Olivera; D Hughes; Gregory M. Pastores; Ramón Nuviala Mateo; J.I. Pérez-Calvo; Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Molecular Genetics and Metabolism 2011, 104, 301-307, 10.1016/j.ymgme.2011.06.021.
- Subroto Chatterjee; Prabodh Gupta; Reed E. Pyeritz; Peter O. Kwiterovich; Immunohistochemical Localization of Giycosphingolipid in Urinary Renal Tubular Cells in Fabry’s Disease. American Journal of Clinical Pathology 1984, 82, 24-28, 10.1093/ajcp/82.1.24.
- Sadanori Nagao; Norio Satoh; Shogo Inaba; Susumu Iijima; CONCENTRIC LAMELLAR SPHERES IN URINE FROM A FEMALE CARRIER OF AND PATIENTS WITH FABRY'S DISEASE. The Journal of Dermatology 1985, 12, 70-78, 10.1111/j.1346-8138.1985.tb01540.x.
- Robert J. Desnick; Glyn Dawson; Susan J. Desnick; Charles C. Sweeley; William Krivit; Diagnosis of Glycosphingolipidoses by Urinary-Sediment Analysis. New England Journal of Medicine 1971, 284, 739-744, 10.1056/nejm197104082841401.
- Mathu Selvarajah; Kathy Nicholls; Tim D. Hewitson; Gavin J. Becker; Targeted urine microscopy in Anderson-Fabry Disease: a cheap, sensitive and specific diagnostic technique. Nephrology Dialysis Transplantation 2011, 26, 3195-3202, 10.1093/ndt/gfr084.
- Linda Van Der Tol; Einar Svarstad; Alberto Ortiz; Camilla Tøndel; João Paulo Oliveira; Liffert Vogt; Stephen Waldek; D Hughes; Robin H Lachmann; Wim Terryn; et al.Carla E. HollakSandrine FlorquinMarius A. Van Den Bergh WeermanChristoph WannerMichael L. WestMarieke BiegstraatenGabor E. LinthorstAlberto Ortiz Chronic kidney disease and an uncertain diagnosis of Fabry disease: Approach to a correct diagnosis. Molecular Genetics and Metabolism 2015, 114, 242-247, 10.1016/j.ymgme.2014.08.007.
- Rannveig Skrunes; Camilla Tøndel; Sabine Leh; Kristin Kampevold Larsen; Gunnar Houge; Einar Skulstad Davidsen; Carla Hollak; André B.P. Van Kuilenburg; Frédéric M. Vaz; Einar Svarstad; et al. Long-Term Dose-Dependent Agalsidase Effects on Kidney Histology in Fabry Disease. Clinical Journal of the American Society of Nephrology 2017, 12, 1470-1479, 10.2215/cjn.01820217.
- Uma Ramaswami; Daniel G. Bichet; Lorne A. Clarke; Gabriela Dostalova; Alejandro Fainboim; Andreas Fellgiebel; Cassiano M. Forcelini; Kristina An Haack; Robert J. Hopkin; Michael Mauer; et al.Behzad NajafianC. Ronald ScottSuma P. ShankarBeth L. ThurbergCamilla TøndelAnna Tylki-SzymańskaBernard BénichouFrits A. Wijburg Low-dose agalsidase beta treatment in male pediatric patients with Fabry disease: A 5-year randomized controlled trial.. Molecular Genetics and Metabolism 2019, 127, 86-94, 10.1016/j.ymgme.2019.03.010.
- Behzad Najafian; Einar Svarstad; Leif Bostad; Marie-Claire Gubler; Camilla Tøndel; Chester Whitley; Michael Mauer; Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney International 2011, 79, 663-670, 10.1038/ki.2010.484.
- Agnes B. Fogo; Leif Bostad; Einar Svarstad; William J. Cook; Solange Moll; Federic Barbey; Laurette Geldenhuys; Michael West; Dusan Ferluga; Bojan Vujkovac; et al.Alexander J. HowieÁine BurnsRoy ReeveStephen WaldekLaure-Hélène NoëlJean-Pierre GrünfeldCarmen ValbuenaJoão Paulo OliveiraJustus MüllerFrank BreunigXiao ZhangDavid G. Warnockall members of the International Study Group of Fabry Nephropathy (ISGFN) Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrology Dialysis Transplantation 2009, 25, 2168-2177, 10.1093/ndt/gfp528.
- Carmen Valbuena; Elísio Carvalho; Manuela Bustorff; Mariana Ganhão; Sandra Relvas; Rosete Nogueira; Fatima Carneiro; João Paulo Oliveira; Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Archiv 2008, 453, 329-338, 10.1007/s00428-008-0653-2.
- Edgar G. Fischer; Michael J Moore; Donna J Lager; Fabry disease: a morphologic study of 11 cases. Modern Pathology 2006, 19, 1295-1301, 10.1038/modpathol.3800634.
- Einar Svarstad; Sabine Leh; Rannveig Skrunes; Kristin Kampevold Larsen; Øystein Eikrem; Camilla Tøndel; Bedside Stereomicroscopy of Fabry Kidney Biopsies: An Easily Available Method for Diagnosis and Assessment of Sphingolipid Deposits. Nephron 2017, 138, 13-21, 10.1159/000479751.
- Beth L. Thurberg; Helmut Rennke; Robert B. Colvin; Steven Dikman; Ronald E. Gordon; A. Bernard Collins; Robert J. Desnick; Michael O'callaghan; Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney International 2002, 62, 1933-1946, 10.1046/j.1523-1755.2002.00675.x.
- Dominique P. Germain; Stephen Waldek; Maryam Banikazemi; David A. Bushinsky; Joel Charrow; Robert J. Desnick; Philip Lee; Thomas Loew; Anouk C. Vedder; Rekha Abichandani; et al.William R. WilcoxNathalie Guffon Sustained, Long-Term Renal Stabilization After 54 Months of Agalsidase β Therapy in Patients with Fabry Disease. Journal of the American Society of Nephrology 2007, 18, 1547-1557, 10.1681/asn.2006080816.
- Uma Ramaswami; Michael Beck; Derralynn Hughes; Christoph Kampmann; Jaco Botha; Guillem Pintos-Morell; Michael L West; Dau-Ming Niu; Kathleen Nicholls; Roberto Giugliani; et al.FOS Study Group Cardio- Renal Outcomes With Long- Term Agalsidase Alfa Enzyme Replacement Therapy: A 10- Year Fabry Outcome Survey (FOS) Analysis.. Drug Design, Development and Therapy 2019, 13, 3705-3715, 10.2147/DDDT.S207856.
- Christoph Wanner; Ulla Feldt-Rasmussen; Ana Jovanovic; Aleš Linhart; Meng Yang; Elvira Ponce; Eva Brand; Dominique P. Germain; Derralynn A. Hughes; John L. Jefferies; et al.Ana Maria MartinsAlbina NowakBojan VujkovacFrank WeidemannMichael L. WestAlberto Ortiz Cardiomyopathy and kidney function in agalsidase beta‐treated female Fabry patients: a pre‐treatment vs. post‐treatment analysis. ESC Heart Failure 2020, 7, 825-834, 10.1002/ehf2.12647.
- Raphael Schiffmann; Hasan Askari; Margaret Timmons; Chevalia Robinson; William Benko; Roscoe O. Brady; Markus Ries; Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing.. Journal of the American Society of Nephrology 2007, 18, 1576-83, 10.1681/ASN.2006111263.
- Christoph Wanner; João P. Oliveira; Alberto Ortiz; Michael Mauer; Dominique P. Germain; Gabor E. Linthorst; Andreas L. Serra; László Maródi; Renzo Mignani; Bruno Cianciaruso; et al.Bojan VujkovacRoberta LemayDana Beitner-JohnsonStephen WaldekDavid G. Warnock Prognostic Indicators of Renal Disease Progression in Adults with Fabry Disease: Natural History Data from the Fabry Registry. Clinical Journal of the American Society of Nephrology 2010, 5, 2220-2228, 10.2215/cjn.04340510.
- Maarten W Taal; B.M. Brenner; Predicting initiation and progression of chronic kidney disease: Developing renal risk scores. Kidney International 2006, 70, 1694-1705, 10.1038/sj.ki.5001794.
- Franc Verovnik; Davorin Benko; Bojan Vujkovac; Gabor E Linthorst; Remarkable variability in renal disease in a large Slovenian family with Fabry disease. European Journal of Human Genetics 2004, 12, 678-681, 10.1038/sj.ejhg.5201184.
- Antonio Junior Lepedda; Laura Fancellu; Elisabetta Zinellu; Pierina De Muro; Gabriele Nieddu; Giovanni Andrea Deiana; Piera Canu; Daniela Concolino; Simona Sestito; Marilena Formato; et al.Gianpietro Sechi Urine Bikunin as a Marker of Renal Impairment in Fabry's Disease. BioMed Research International 2013, 2013, 1-9, 10.1155/2013/205948.
- Patrício Aguiar; Olga Azevedo; Rui Pinto; Jacira Marino; Robert Baker; Carlos Cardoso; José Luís Ducla Soares; Derralynn Hughes; New biomarkers defining a novel early stage of Fabry nephropathy: A diagnostic test study. Molecular Genetics and Metabolism 2017, 121, 162-169, 10.1016/j.ymgme.2017.05.007.
- Raphael Schiffmann; Stephen Waldek; Ariela Benigni; Christiane Auray-Blais; Biomarkers of Fabry Disease Nephropathy. Clinical Journal of the American Society of Nephrology 2009, 5, 360-364, 10.2215/cjn.06090809.
- Thaneas Prabakaran; Henrik Birn; B. M. Bibby; Axel Regeniter; Søren S. Sørensen; Ulla Feldt-Rasmussen; Rikke Nielsen; Erik I. Christensen; Long-term enzyme replacement therapy is associated with reduced proteinuria and preserved proximal tubular function in women with Fabry disease. Nephrology Dialysis Transplantation 2013, 29, 619-625, 10.1093/ndt/gft452.
- P. Vylet’Al; H. Hůlková; M. Živná; L. Berná; P. Novák; M. Elleder; S. Kmoch; Abnormal expression and processing of uromodulin in Fabry disease reflects tubular cell storage alteration and is reversible by enzyme replacement therapy. Journal of Inherited Metabolic Disease 2008, 31, 508-517, 10.1007/s10545-008-0900-3.
- Camilla Tøndel; Takahiro Kanai; Kristin Kampevold Larsen; Shuichi Ito; Juan Manuel Politei; David G. Warnock; Einar Svarstad; Foot Process Effacement Is an Early Marker of Nephropathy in Young Classic Fabry Patients without Albuminuria. Nephron 2014, 129, 16-21, 10.1159/000369309.
- Brent Fall; C. Ronald Scott; Michael Mauer; Stuart Shankland; Jeffrey Pippin; Jonathan A. Jefferson; Eric Wallace; David Warnock; Behzad Najafian; Urinary Podocyte Loss Is Increased in Patients with Fabry Disease and Correlates with Clinical Severity of Fabry Nephropathy. PLOS ONE 2016, 11, e0168346, 10.1371/journal.pone.0168346.
- Behzad Najafian; Camilla Tøndel; Einar Svarstad; Marie-Claire Gubler; João-Paulo Oliveira; Michael Mauer; Accumulation of Globotriaosylceramide in Podocytes in Fabry Nephropathy Is Associated with Progressive Podocyte Loss. Journal of the American Society of Nephrology 2020, 31, 865-875, 10.1681/asn.2019050497.
- Hernán Trimarchi; Romina Canzonieri; Cristian Costales-Collaguazo; Juan Politei; Anibal Stern; Matias Paulero; Ivan Gonzalez Hoyos; Amalia Schiel; Tatiana Rengel; Mariano Forrester; et al.Fernando LombiVanesa PomeranzRomina IriarteAlexis MuryanElsa Zotta Early decrease in the podocalyxin to synaptopodin ratio in urinary Fabry podocytes.. Clinical Kidney Journal 2018, 12, 53-60, 10.1093/ckj/sfy053.
- Carl James May; Moin Saleem; Gavin Iain Welsh; Podocyte Dedifferentiation: A Specialized Process for a Specialized Cell. Frontiers in Endocrinology 2014, 5, 148, 10.3389/fendo.2014.00148.
- Frank Weidemann; Maria Dolores Sanchez-Niño; Juan M. Politei; João Paulo Oliveira; Chistoph Wanner; David G. Warnock; Alberto Ortiz; Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet Journal of Rare Diseases 2013, 8, 116-116, 10.1186/1750-1172-8-116.
- Ester Miranda Pereira; Adalberto Socorro Da Silva; Anatália Labilloy; José Tiburcio Do Monte Neto; Semiramis Jamil Hadad Do Monte; Podocyturia in Fabry disease. Brazilian Journal of Nephrology 2016, 38, 49-53, 10.5935/0101-2800.20160008.
- Helen M. Colhoun; M. Loredana Marcovecchio; Biomarkers of diabetic kidney disease. Diabetologia 2018, 61, 996-1011, 10.1007/s00125-018-4567-5.
- Yong-Ho Lee; Heejung Bang; Dae Jung Kim; How to Establish Clinical Prediction Models. Endocrinology and Metabolism 2016, 31, 38-44, 10.3803/enm.2016.31.1.38.
- Helen M. Colhoun; M. Loredana Marcovecchio; Biomarkers of diabetic kidney disease. Diabetologia 2018, 61, 996-1011, 10.1007/s00125-018-4567-5.
- Yong-Ho Lee; Heejung Bang; Dae Jung Kim; How to Establish Clinical Prediction Models. Endocrinology and Metabolism 2016, 31, 38-44, 10.3803/enm.2016.31.1.38.


