Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Krzysztof Bednarz | -- | 2804 | 2022-04-20 22:17:29 | | | |
| 2 | Rita Xu | Meta information modification | 2804 | 2022-04-21 06:09:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bednarz, K.; Kowalczyk, K.; , .; Czapla, D.; Czarkowski, W.; Kmita, D.; Madej, P. Insulin Resistance in Polycystic Ovary Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/22047 (accessed on 13 June 2026).
Bednarz K, Kowalczyk K, , Czapla D, Czarkowski W, Kmita D, et al. Insulin Resistance in Polycystic Ovary Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/22047. Accessed June 13, 2026.
Bednarz, Krzysztof, Karolina Kowalczyk, , Dominika Czapla, Wiktor Czarkowski, Dominika Kmita, Pawel Madej. "Insulin Resistance in Polycystic Ovary Syndrome" Encyclopedia, https://encyclopedia.pub/entry/22047 (accessed June 13, 2026).
Bednarz, K., Kowalczyk, K., , ., Czapla, D., Czarkowski, W., Kmita, D., & Madej, P. (2022, April 20). Insulin Resistance in Polycystic Ovary Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/22047
Bednarz, Krzysztof, et al. "Insulin Resistance in Polycystic Ovary Syndrome." Encyclopedia. Web. 20 April, 2022.
Copy Citation
Insulin resistance is documented in clamp studies in 75% of women with polycystic ovary syndrome (PCOS). Although it is not included in the diagnostic criteria of PCOS, there is a crucial role of this metabolic impairment, which along with hormonal abnormalities, increase each other in a vicious circle of PCOS pathogenesis. Insulin resistance in this group of patients results from defects at the molecular level, including impaired insulin receptor-related signaling pathways enhanced by obesity and its features: Excess visceral fat, chronic inflammation, and reactive oxygen species.
polycystic ovary syndrome (PCOS)
insulin resistance (IR)
glucagon-like peptide-1 receptor agonists (GLP-1RAs)
1. Introduction
Polycystic ovary syndrome (PCOS) is a significant health burden that impairs women’s quality of life [1]. It is a common condition [1], but its precise worldwide presence is very difficult to evaluate due to the lack of a universal definition for this disease, diversity of its phenotypes, the possible influence of age, ethnicity [2], and different diagnostic criteria usage [3][4]. For these reasons, the prevalence of PCOS ranges from 2.2% to 26% in different studies [5]. Although the exact reason for PCOS is not yet discovered, there are multiple genetic [3][6] and environmental factors that play an important role in the occurrence of this disorder [7][8].
The essence of PCOS is a heterogenous presentation of androgen excess (which appears as hyperandrogenism, hirsutism, acne, alopecia, and seborrhoea), ovulatory dysfunction (which appears as oligo-ovulation, menstrual dysfunction, and subfertility) and polycystic ovarian morphology (PCOM). It is believed that hyperinsulinemia is the main reason for increased androgen secretion because insulin acts as a co-gonadotropin on the ovary, facilitates androgen secretion from the adrenal glands, and modulates releasing of luteinizing hormone (LH) [8]. Because of these reasons, women with increased insulin levels (regardless of their endo-or exogenous origin) have increased prevalence of PCOS. Androgen excess by favoring visceral adiposity contributes to insulin resistance (IR) and to the development of hyperinsulinism [9]. As studies show, visceral adipose tissue in women with PCOS differs from that in healthy women, and in genomic, transcriptomic, and proteomic profiles is like male visceral adipose tissue. However, the fact that PCOS does not appear in every woman with IR or hyperinsulinemia and the fact that not every woman suffering from PCOS presents IR, suggests that there must be another malfunction that favors androgen excess in response to insulin or other triggering factors [8].
Depending on the presenting features, there are three different clinical manifestations of PCOS. The classic phenotype consists of hyperandrogenism and oligo-ovulation. It is linked with the most severe insulin resistance and metabolic comorbidities. The next phenotype is ovulatory PCOS which comprises hyperandrogenism and PCOM. It co-occurs with moderate IR and metabolic dysfunction. The least severe is the non-hyperandrogenic phenotype, which presents with oligo-ovulation, PCOM and has the weakest connection with IR and metabolic comorbidities [8].
Treatment of PCOS should be adjusted to the individual needs of the patient’s complaints and symptoms [10]. Androgen excess, IR, obesity, and oligoovulation might be the targets of treatment [10]. Early prevention of metabolic consequences needs to be highlighted. Hyperglycemia, obesity, hypertension, and dyslipidemia define the metabolic syndrome, which occurs with PCOS more often [11]. Therefore, women with PCOS must be regularly screened for obesity (using BMI and waist circumference), blood pressure, glucose, and lipid profile [12]. The excess nutrients cause hypertrophy or hyperplasia of adipocyte and induces IR, hyperinsulinemia [11], higher serum free fatty acids, lipogenesis, and increased fat storage in the liver, pancreas, and skeletal muscles [13]. In consequence, glucose tolerance disorders lead to type-2 diabetes mellitus (T2DM) [12].
Education, counseling, and a healthy lifestyle, including appropriate diets and physical activity, should be the focus of the therapeutic proceedings [1][14]. A low-glycemic load (20 to 40 g/day), as well as low-fat diets (40 g of fat per day), were confirmed to be effective for weight control but not for biochemical hyperandrogenism. There is also no indication that a low-glycemic loan diet was better than a low-fat diet for attenuating hyperandrogenism [15]. There is not enough strong evidence that any specific diet leads to better clinical outcomes. Diet should be managed individually and should include energy deficit to reduce weight. Regular healthy eating and physical exercise improve general health conditions and optimize hormonal balance [1]. Losing 5–10% of body weight is considered a significant clinical improvement, especially among patients with excess weight [1]. A minimum of 250 min/week of moderate intensity or 150 min/week of vigorous intensity activities or an equivalent combination of both, and muscle-strengthening activities involving major muscle groups on two non-consecutive days/week are recommended [1]. However, among obese women, joints problem and arthritis could be significant limitations to exercise. Therefore, physical activity must be tailored to personal abilities [16]. Lifestyle interventions should be supported by interactions with a mental health professional and should include psychological care to improve a patient’s emotional state. It is worth noting that anxiety and depression relatively often coexist with PCOS. The research confirmed depressive symptoms in 44% and anxiety disorder in 41.9% of women with PCOS [1]. Lower mood, loss of motivation co-occurring with negative body image usually causes the lifestyle change to be a real challenge for patients. Women with PCOS are more likely to follow weight management practices [17], although they also consider achieving and maintaining this reduction challenging [18]. Moreover, there is evidence that suggests that women with PCOS may encounter more barriers and difficulties in losing weight due to reduced utilization of lipid stores [19] and decreased brown adipose tissue activity [20]. There is also a lack of data describing long-term compliance among patients with PCOS with the dietary regime and lifestyle modifications, which translates into the effectiveness of the therapy. Although even in well-supported programs such as lifestyle intervention in randomized controlled trials, there was a high risk of dropping out during long-term follow-up [21], and investigators report high attrition rates [22][23]. It is considered that among adolescents with PCOS, only around 60% adhere to nutritional interventions [24].
On the other hand, more radical methods of treatment, like bariatric surgery and specific medicaments, can be considered. According to guidelines for obesity management [25], bariatric surgery is recommended for patients with BMI ≥ 35 kg/m2 and a minimum of one severe complication. Evidence in the treatment of PCOS is limited, and there is no recommendation available. This experimental therapy can induce malabsorption and eating disorders. It could also impact future pregnancies and the fetus, and it increases neonatal mortality [26]. Anti-obesity medications can be a consideration for adults with PCOS. However, due to the possible side effects, as well as variable availability, it is recommended to avoid pregnancy as long as such medications are being taken [1]. Reduction of obesity, as well as testosterone levels, and as a result, improving ovulation, can be achieved by using metformin. It decreases gluconeogenesis, lipogenesis, and enhances uptake of glucose in the skeletal muscle, liver, and adipose tissue. It should be recommended in PCOS with BMI ≥ 25 kg/m2 in addition to a healthy lifestyle [27], particularly for patients with disturbed glucose tolerance [28]. Different diabetic medicines like the insulin-sensitizing treatment in PCOS-glucagon-like peptide-1 receptor agonists (GLP-1RAs), dipeptidyl peptidase 4 (DPP-4) inhibitors, and sodium-glucose cotransporter (SGLT2) inhibitors could also be considered as a part of the treatment of PCOS [29][30].
GLP-1RAs release glucose-dependent insulin from the pancreatic cells. The main action of GLP-1RAs is summarized in the diagram (Figure 1). Using GLP-1RAs leads to a reduction in weight, testosterone levels, and improves ovulation among obese patients [31]. The entry is focused on the role of the GLP-1RAs in the treatment of PCOS. The entry presents current data on the possible use of GLP-1 agonists in PCOS therapy, based on articles from 2010 to 2022.

Figure 1. Effects of GLP-1RAs on organs and tissues. The drugs increase glucose-dependent insulin release by acting on pancreatic islets β cells and inhibit glucagon release by acting on pancreatic islets α cells. It leads to decreased glucose release from liver glycogen to circulation. They also enhance the proliferation of β cells. By acting on the central nervous system, GLP-1RAs reduce appetite, which along with slower gastric emptying, leads to reduced food intake. All these effects contribute to a reduction in blood glucose levels. Pictures used to create this figure is designed by macrovector Freepik.
2. The Pathomechanism and Role of IR in PCOS
IR is an impairment of insulin effect on cells, especially muscle cells, adipocytes, and hepatocytes. It leads to carbohydrate, lipid, and protein metabolism alterations. IR is an important element of metabolic disorders, including T2DM and pre-diabetes (impaired glucose tolerance and impaired fasting glucose) [32]. In PCOS, IR is present in obese and overweight patients, as well as in those of normal weight, although in the latter group, with a lower frequency (59.3% in normal weight vs. 77.5% in overweight and 93.9% in obese patients) [33]. This difference is probably the result of an increase in adipose tissue volume, which plays a crucial role in the pathomechanism of IR. The incidence of IR is also different according to the phenotype of PCOS. It has been shown that IR is present more often in the classic phenotype (80.4%) than in the ovulatory (65.0%) and normoandrogenic (38.1%) [34]. Moreover, it is more frequent in patients with anovulatory compared to those with ovulatory cycles in PCOS [35].
The mechanism of IR in PCOS is complex and still not fully understood. Increased adipose tissue volume, especially visceral adipose tissue, plays an important role in IR development [9]. It is influenced by many factors, including excessive energy intake and high androgen levels that promote a change in body fat distribution with an increase in visceral fat volume [9][36]. In PCOS, hypertrophic adipocytes are associated with metabolic disorders [37]. In a study on adipocytes from healthy women, it was observed that testosterone, acting through its receptor, induces IR in these cells by affecting insulin-stimulated phosphorylation of protein kinase C (PKC) (Figure 2) [38].

Figure 2. Alterations in the insulin receptor pathway, resulting from the activity of pro-inflammatory cytokines and androgens. Tumour necrosis factor α (TNF-α) affects insulin signaling by phosphorylation of serine in insulin receptor substrate-1 (IRS-1) through activation of several serine kinases, including c-Jun-NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK). It inhibits insulin-induced tyrosine phosphorylation of IRS-1 and downregulates phosphoinositide 3-kinase (PI3K) activity. Decreased adiponectin concentration results in increased membrane sn-1,2-diacylglycerols (sn-1,2-DAGs) activity. It leads to impaired kinases activity and decreased insulin signaling. Testosterone induces insulin resistance in cells by affecting insulin-stimulated phosphorylation of protein kinase C (PKC). All these mechanisms contribute to decreased glucose transporter type 4 (GLUT-4) expression and decreased glucose transport into cells.
In over-expanded adipose tissue, the profile of adipokines secreted by adipocytes is impaired. As adipose tissue volume increases, the concentration of adiponectin, having an insulin-sensitizing and anti-inflammatory impact, declines. Its lower concentration is associated with the development of IR and T2DM [39]. Adiponectin reduces lipid storage in the liver and muscles, which results in decreased plasma membrane sn-1,2-diacylglycerol-induced PKC activity and increases insulin signaling. Adiponectin mediates these effects by promoting the storage of triglyceride (TG) in white adipose tissue through stimulation of lipoprotein lipase and by increasing muscle fat oxidation and glucose uptake by stimulation of 5′ adenosine monophosphate-activated protein kinase (AMPK) in muscle (Figure 3) [40]. Moreover, in PCOS, adiponectin levels are decreased in both obese and nonobese patients [41][42]. Changes in other adipokines, such as apelin, vaspin, resistin, and chemerin, are also observed in women with PCOS. Apelin is adipokine expressed in adipocytes, and ovarian cells, and its expression is stimulated by insulin. It increases the secretion of progesterone and estradiol in granulosa cells in response to stimulation by insulin-like growth factor-1 (IGF-1). Vaspin, an adipokine expressed mainly in visceral adipose tissue and in the ovary, enhances granulosa cells’ steroidogenesis and proliferation. The expression of resistin is upregulated by androgens, therefore there is a positive association between hyperandrogenism and high levels of resistin in PCOS patients. Resistin may be involved in the pathogenesis of IR in PCOS. Chemerin is expressed in ovarian cells and plays an important role in adipose tissue inflammation [43].
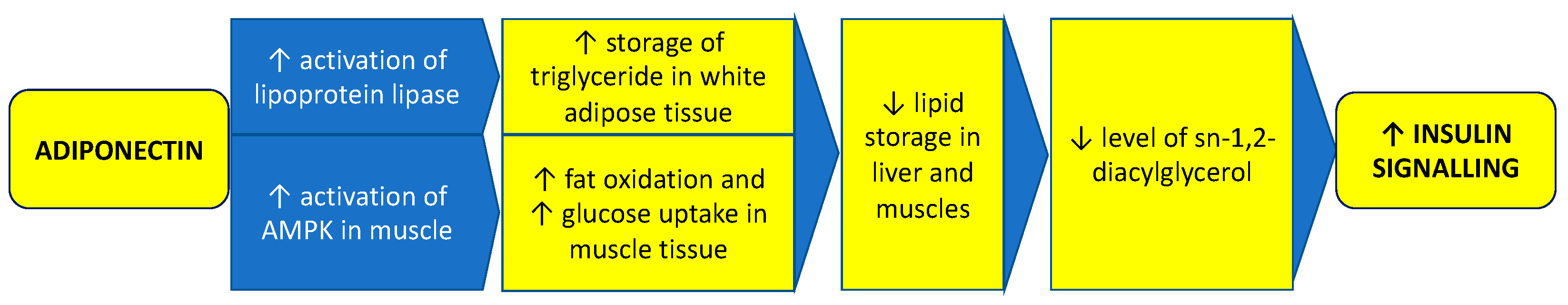
Figure 3. Role of adiponectin in maintaining proper insulin signaling. Adiponectin reduces lipid storage in the liver and muscles, which results in decreased membrane sn-1,2-diacylglycerol levels and increases insulin signaling. Adiponectin mediates these effects by promoting the storage of triglyceride in white adipose tissue through stimulation of lipoprotein lipase and by increasing muscle fat oxidation and glucose uptake by stimulation of 5′ adenosine monophosphate-activated protein kinase (AMPK) in muscle.
Adipose tissue growth without adequate vascularization results in hypoxia. Reduced adipose tissue oxygenation contributes to IR by decreasing branched-chain amino acid catabolism and increasing plasma branched-chain amino acid concentrations [44]. Chronic inflammation also develops with increased levels of tumor necrosis factor α (TNF-α), monocyte chemotactic protein-1 (MCP-1), C-reactive protein (CRP), interleukin-1 (IL-1), interleukin-6 (IL-6), and other pro-inflammatory cytokines, which have been found elevated in PCOS. Additionally, hyperandrogenism can increase inflammation through activation of the nuclear factor kappa B (NF-κB) inflammation pathway [45][46]. Hormones, adipokines, and cytokines associated with inflammation affect the intracellular enzymatic pathways associated with the insulin receptor. Insulin acts through binding and activation of insulin receptors type A and B (IR-A and IR-B) located in the cell membrane. Activation of receptor subunits, which are glycoproteins with tyrosine kinase activity, leads to phosphorylation of the insulin receptor substrates (IRS-1 and IRS-2). Then, the signal is transmitted through intracellular signalling pathways. The phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway is involved in carbohydrate metabolism by increasing intracellular glucose transport through glucose transporter type 4 (GLUT-4) in muscle cells and adipocytes, inhibiting hepatic gluconeogenesis and glycogenolysis, and promoting glycogenesis. The mitogen-activated protein kinase (MAPK) pathway is involved in promoting proliferation and cell growth [47]. Defects in insulin signal transduction play a crucial role in the pathomechanism of IR. It is a result of protein modifications, such as increased IRS-1 serine phosphorylation, which affects metabolic pathways in many tissues, including the ovary [48]. TNF-α affects insulin signaling by phosphorylation of serine in IRS-1 through activation of several serine kinases, including c-Jun-NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK), which inhibits insulin-induced tyrosine phosphorylation of IRS-1 and downregulates PI3K activity [49]. It causes decreased GLUT-4 expression and decreased glucose transport into cells (Figure 2). Moreover, mitochondrial damage, increased oxidative stress, and endoplasmic reticulum stress are also involved in the development of IR [50]. Metabolic inflexibility is the impaired ability to switch from lipid to carbohydrate oxidation under insulin-stimulated conditions. This state is associated with IR. However, some data show that PCOS women have normal metabolic flexibility, which could suggest a distinct pathophysiological mechanism for IR in this group [51].
Studies on mice have shown that activation of androgen receptors by dihydrotestosterone in the brains of the tested animals led to the development of IR, whereas rodents deprived of these receptors did not develop this type of disorder. That observation suggests that androgens may also promote IR by acting on the central nervous system [52]. Moreover, prenatal exposure to high levels of androgens can probably lead to the development of IR and PCOS [53][54][55].
The role of other factors in pathomechanism of IR is also under investigation. MicroRNA (miRNA) are about 22 nucleotides long non-coding RNA molecules that are responsible for post-transcriptional regulation of gene expression [56]. Studies show that these particles, located in exosomes of adipocytes and macrophages of adipose tissue and secreted from them into the circulation, can influence the development of IR through mechanisms, such as inhibition of peroxisome proliferator-activated receptor (PPAR-gamma) expression and affecting PI3K/AKT-GLUT4 signaling pathway [57][58][59]. An altered profile of miRNA particles in plasma has been observed in PCOS [60]. As a result, miRNAs can be used as diagnostic markers and, in the future, be important in the therapy of PCOS and IR.
The result of all the processes described above is impaired glucose transport into cells by insulin-dependent membrane GLUT 4 transporters, reduced glycogen synthesis and decreased inhibition of hepatic gluconeogenesis. This leads to hyperglycemia and to compensatory hyperinsulinemia. IR, the resulting hyperinsulinemia, and increased androgen levels influence each other, and it is difficult to determine which phenomenon occurs first (Figure 4) [61]. Insulin acting on ovarian theca cells increases androgen synthesis by increasing activity of the cytochrome P450C17α, which has 17-hydroxylase and 17,20-lyase activities [62]. Insulin acts on ovarian theca cells by further increasing LH stimulatory effect [63]. Moreover, insulin affects the hypothalamic-pituitary system. Through MAPK and increased gonadotropin-releasing hormone (GnRH) gene expression, it increases the frequency and amplitude of GnRH release pulses and, in consequence, LH release, which stimulates ovarian cells to androgen synthesis [64]. Insulin also upregulates adrenal androgen synthesis by affecting steroidogenic factor 1 (SF-1), which is a transcriptional factor playing an important role in steroid hormone synthesis by regulating the transcription of steroidogenic genes, including StAR, Cyp11a1, Cyp17, Cyp11b1, Cyp11b2, and 3β-Hsd [65]. Moreover, insulin decreases the release of sex hormone-binding globulin (SHBG) from the liver, thus increasing the amount of the free, non-protein-bound androgen fraction, which exerts biological effects on tissues. Studies indicate that SHBG can be useful as a marker for a higher risk of PCOS [66].

Figure 4. Possible relationships between polycystic ovary syndrome and insulin resistance. The main conclusion is that insulin resistance, the resulting hyperinsulinemia, and increased androgen levels influence each other, and it is difficult to determine which phenomenon occurs first.
References
- Misso, M.; Costello, M.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R. International Evidencebased for the Assessment and management of Polycystic Ovary Syndrome; Monash University: Melbourne, Australia, 2018; Volume 2.
- Miazgowski, T.; Martopullo, I.; Widecka, J.; Miazgowski, B.; Brodowska, A. National and Regional Trends in the Prevalence of Polycystic Ovary Syndrome since 1990 within Europe: The Modeled Estimates from the Global Burden of Disease Study 2016. Arch. Med. Sci. 2021, 17, 343–351.
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C. The Prevalence of Polycystic Ovary Syndrome: A Brief Systematic Review. J. Hum. Reprod. Sci. 2020, 13, 261.
- Sirmans, S.; Pate, K. Epidemiology, Diagnosis, and Management of Polycystic Ovary Syndrome. Clin. Epidemiol. 2013, 6, 1–13.
- Tehrani, F.; Simbar, M.; Tohidi, M.; Hosseinpanah, F.; Azizi, F. The Prevalence of Polycystic Ovary Syndrome in a Community Sample of Iranian Population: Iranian PCOS Prevalence Study. Reprod. Biol. Endocrinol. 2011, 9, 39.
- Jones, M.R.; Goodarzi, M.O. Genetic Determinants of Polycystic Ovary Syndrome: Progress and Future Directions. Fertil. Steril. 2016, 106, 25–32.
- Merkin, S.S.; Phy, J.L.; Sites, C.K.; Yang, D. Environmental Determinants of Polycystic Ovary Syndrome. Fertil. Steril. 2016, 106, 16–24.
- Escobar-Morreale, H.F. Polycystic Ovary Syndrome: Definition, Aetiology, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284.
- Polak, A.M.; Adamska, A.; Krentowska, A.; Łebkowska, A.; Hryniewicka, J.; Adamski, M.; Kowalska, I. Body Composition, Serum Concentrations of Androgens and Insulin Resistance in Different Polycystic Ovary Syndrome Phenotypes. J. Clin. Med. 2020, 9, 732.
- Conway, G.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Franks, S.; Gambineri, A.; Kelestimur, F.; Macut, D.; Micic, D.; Pasquali, R.; et al. The Polycystic Ovary Syndrome: A Position Statement from the European Society of Endocrinology. Eur. J. Endocrinol. 2014, 171, P1–P29.
- Witchel, S.F.; Oberfield, S.E.; Peña, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment with Emphasis on Adolescent Girls. J. Endocr. Soc. 2019, 3, 1545–1573.
- Rasquin Leon, L.I.; Anastasopoulou, C.; Mayrin, J.V. Polycystic Ovarian Disease; StatPearls: Treasure Island, FL, USA, 2022.
- Samuel, V.T.; Shulman, G.I. The Pathogenesis of Insulin Resistance: Integrating Signaling Pathways and Substrate Flux. J. Clin. Investig. Am. Soc. Clin. Investig. 2016, 126, 12–22.
- Ells, L.J.; Rees, K.; Brown, T.; Mead, E.; Al-Khudairy, L.; Azevedo, L.; McGeechan, G.J.; Baur, L.; Loveman, E.; Clements, H.; et al. Interventions for Treating Children and Adolescents with Overweight and Obesity: An Overview of Cochrane Reviews. Int. J. Obes. 2018, 42, 1823–1833.
- Wong, J.M.W.; Gallagher, M.; Gooding, H.; Feldman, H.A.; Gordon, C.M.; Ludwig, D.S.; Ebbeling, C.B. A Randomized Pilot Study of Dietary Treatments for Polycystic Ovary Syndrome in Adolescents. Pediatr. Obes. 2016, 11, 210–220.
- Legro, R. Obesity and PCOS: Implications for Diagnosis and Treatment. Semin. Reprod. Med. 2012, 30, 496–506.
- Moran, L.J.; Brown, W.J.; McNaughton, S.A.; Joham, A.E.; Teede, H.J. Weight Management Practices Associated with PCOS and Their Relationships with Diet and Physical Activity. Hum. Reprod. 2017, 32, 669–678.
- Lim, S.; Smith, C.A.; Costello, M.F.; MacMillan, F.; Moran, L.; Ee, C. Barriers and Facilitators to Weight Management in Overweight and Obese Women Living in Australia with PCOS: A Qualitative Study. BMC Endocr. Disord. 2019, 19, 106.
- Whigham, L.; Butz, D.; Dashti, H.; Tonelli, M.; Johnson, L.; Cook, M.; Porter, W.; Eghbalnia, H.; Markley, J.; Lindheim, S.; et al. Metabolic Evidence of Diminished Lipid Oxidation in Women with Polycystic Ovary Syndrome. Curr. Metab. 2014, 1, 269–278.
- Shorakae, S.; Jona, E.; de Courten, B.; Lambert, G.W.; Lambert, E.A.; Phillips, S.E.; Clarke, I.J.; Teede, H.J.; Henry, B.A. Brown Adipose Tissue Thermogenesis in Polycystic Ovary Syndrome. Clin. Endocrinol. 2019, 90, 425–432.
- Domecq, J.P.; Prutsky, G.; Mullan, R.J.; Hazem, A.; Sundaresh, V.; Elamin, M.B.; Phung, O.J.; Wang, A.; Hoeger, K.; Pasquali, R.; et al. Lifestyle Modification Programs in Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2013, 98, 4655–4663.
- Kim, C.-H.; Lee, S.-H. Effectiveness of Lifestyle Modification in Polycystic Ovary Syndrome Patients with Obesity: A Systematic Review and Meta-Analysis. Life 2022, 12, 308.
- Kazemi, M.; McBreairty, L.E.; Zello, G.A.; Pierson, R.A.; Gordon, J.J.; Serrao, S.B.; Chilibeck, P.D.; Chizen, D.R. A Pulse-Based Diet and the Therapeutic Lifestyle Changes Diet in Combination with Health Counseling and Exercise Improve Health-Related Quality of Life in Women with Polycystic Ovary Syndrome: Secondary Analysis of a Randomized Controlled Trial. J. Psychosom. Obstet. Gynecol. 2020, 41, 144–153.
- Carolo, A.L.; Mendes, M.C.; Rosa e Silva, A.C.J.D.S.; Vieira, C.S.; de Sá, M.F.S.; Ferriani, R.A.; dos Reis, R.M. O Aconselhamento Nutricional Promove Mudanças Nos Hábitos Alimentares de Adolescentes Com Excesso de Peso e Obesas e Com Síndrome Dos Ovários Policísticos. Rev. Bras. Ginecol. Obstet. 2017, 39, 692–696.
- Scottish Intercollegiate Guidelines Network. Scottish Intercollegiate Guidelines Network Part of NHS Quality Improvement Scotland SIGN Management of Obesity; The Scottish Intercollegiate Guidelines Network (SIGN): Edinburgh, UK, 2010.
- Johansson, K.; Cnattingius, S.; Näslund, I.; Roos, N.; Trolle Lagerros, Y.; Granath, F.; Stephansson, O.; Neovius, M. Outcomes of Pregnancy after Bariatric Surgery. N. Engl. J. Med. 2015, 372, 814–824.
- Naderpoor, N.; Shorakae, S.; de Courten, B.; Misso, M.L.; Moran, L.J.; Teede, H.J. Metformin and Lifestyle Modification in Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. Hum. Reprod. Update 2015, 21, 560–574.
- Wild, R.A.; Carmina, E.; Diamanti-Kandarakis, E.; Dokras, A.; Escobar-Morreale, H.F.; Futterweit, W.; Lobo, R.; Norman, R.J.; Talbott, E.; Dumesic, D.A. Assessment of Cardiovascular Risk and Prevention of Cardiovascular Disease in Women with the Polycystic Ovary Syndrome: A Consensus Statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J. Clin. Endocrinol. Metab. 2010, 95, 2038–2049.
- Devin, J.K.; Nian, H.; Celedonio, J.E.; Wright, P.; Brown, N.J. Sitagliptin Decreases Visceral Fat and Blood Glucose in Women with Polycystic Ovarian Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, 136–151.
- Javed, Z.; Papageorgiou, M.; Deshmukh, H.; Rigby, A.S.; Qamar, U.; Abbas, J.; Khan, A.Y.; Kilpatrick, E.S.; Atkin, S.L.; Sathyapalan, T. Effects of Empagliflozin on Metabolic Parameters in Polycystic Ovary Syndrome: A Randomized Controlled Study. Clin. Endocrinol. 2019, 90, 805–813.
- Niafar, M.; Pourafkari, L.; Porhomayon, J.; Nader, N. A Systematic Review of GLP-1 Agonists on the Metabolic Syndrome in Women with Polycystic Ovaries. Arch. Gynecol. Obstet. 2016, 293, 509–515.
- Czech, M.P. Mechanisms of Insulin Resistance Related to White, Beige, and Brown Adipocytes. Mol. Metab. 2020, 34, 27–42.
- Tosi, F.; Bonora, E.; Moghetti, P. Insulin Resistance in a Large Cohort of Women with Polycystic Ovary Syndrome: A Comparison between Euglycaemic-Hyperinsulinaemic Clamp and Surrogate Indexes. Hum. Reprod. 2017, 32, 2515–2521.
- Moghetti, P.; Tosi, F.; Bonin, C.; di Sarra, D.; Fiers, T.; Kaufman, J.M.; Giagulli, V.A.; Signori, C.; Zambotti, F.; Dall’Alda, M.; et al. Divergences in Insulin Resistance between the Different Phenotypes of the Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E628–E637.
- Cho, L.W.; Kilpatrick, E.S.; Keevil, B.G.; Jayagopal, V.; Coady, A.M.; Rigby, A.S.; Atkin, S.L. Insulin Resistance Variability in Women with Anovulatory and Ovulatory Polycystic Ovary Syndrome, and Normal Controls. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2011, 43, 141–145.
- Borruel, S.; Fernández-Durán, E.; Alpañés, M.; Martí, D.; Álvarez-Blasco, F.; Luque-Ramírez, M.; Escobar-Morreale, H.F. Global Adiposity and Thickness of Intraperitoneal and Mesenteric Adipose Tissue Depots Are Increased in Women with Polycystic Ovary Syndrome (PCOS). J. Clin. Endocrinol. Metab. 2013, 98, 1254–1263.
- Mannerås-Holm, L.; Leonhardt, H.; Kullberg, J.; Jennische, E.; Odén, A.; Holm, G.; Hellström, M.; Lönn, L.; Olivecrona, G.; Stener-Victorin, E.; et al. Adipose Tissue Has Aberrant Morphology and Function in PCOS: Enlarged Adipocytes and Low Serum Adiponectin, but Not Circulating Sex Steroids, Are Strongly Associated with Insulin Resistance. J. Clin. Endocrinol. Metab. 2011, 96, E304–E311.
- Corbould, A. Chronic Testosterone Treatment Induces Selective Insulin Resistance in Subcutaneous Adipocytes of Women. J. Endocrinol. 2007, 192, 585–594.
- Liu, C.; Feng, X.; Li, Q.; Wang, Y.; Li, Q.; Hua, M. Adiponectin, TNF-α and Inflammatory Cytokines and Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. Cytokine 2016, 86, 100–109.
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by Which Adiponectin Reverses High Fat Diet-Induced Insulin Resistance in Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593.
- Li, S.; Huang, X.; Zhong, H.; Peng, Q.; Chen, S.; Xie, Y.; Qin, X.; Qin, A. Low Circulating Adiponectin Levels in Women with Polycystic Ovary Syndrome: An Updated Meta-Analysis. Tumour Biol. 2014, 35, 3961–3973.
- Lin, K.; Sun, X.; Wang, X.; Wang, H.; Chen, X. Circulating Adipokine Levels in Nonobese Women with Polycystic Ovary Syndrome and in Nonobese Control Women: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2021, 11, 537809.
- Mehrabani, S.; Arab, A.; Karimi, E.; Nouri, M.; Mansourian, M. Blood Circulating Levels of Adipokines in Polycystic Ovary Syndrome Patients: A Systematic Review and Meta-Analysis. Reprod. Sci. 2021, 28, 3032–3050.
- Cifarelli, V.; Beeman, S.C.; Smith, G.I.; Yoshino, J.; Morozov, D.; Beals, J.W.; Kayser, B.D.; Watrous, J.D.; Jain, M.; Patterson, B.W.; et al. Decreased Adipose Tissue Oxygenation Associates with Insulin Resistance in Individuals with Obesity. J. Clin. Investig. 2020, 130, 6688–6699.
- González, F.; Sreekumaran Nair, K.; Daniels, J.K.; Basal, E.; Schimke, J.M. Hyperandrogenism Sensitizes Mononuclear Cells to Promote Glucose-Induced Inflammation in Lean Reproductive-Age Women. Am. J. Physiol. Endocrinol. Metab. 2012, 302, 297–306.
- González, F.; Rote, N.S.; Minium, J.; Weaver, A.L.; Kirwan, J.P. Elevated Circulating Levels of Macrophage Migration Inhibitory Factor in Polycystic Ovary Syndrome. Cytokine 2010, 51, 240–244.
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44.
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome Revisited: An Update on Mechanisms and Implications. Endocr. Rev. 2012, 33, 981–1030.
- Shibata, T.; Takaguri, A.; Ichihara, K.; Satoh, K. Inhibition of the TNF-α-Induced Serine Phosphorylation of IRS-1 at 636/639 by AICAR. J. Pharmacol. Sci. 2013, 122, 93–102.
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell Physiol. 2019, 234, 8152–8161.
- Adamska, A.; Karczewska-Kupczewska, M.; Nikołajuk, A.; Otziomek, E.; Górska, M.; Kowalska, I.; Straczkowski, M. Normal Metabolic Flexibility despite Insulin Resistance Women with Polycystic Ovary Syndrome. Endocr. J. 2013, 60, 1107–1113.
- Navarro, G.; Allard, C.; Morford, J.J.; Xu, W.; Liu, S.; Molinas, A.J.; Butcher, S.M.; Fine, N.H.; Blandino-Rosano, M.; Sure, V.N.; et al. Androgen Excess in Pancreatic β Cells and Neurons Predisposes Female Mice to Type 2 Diabetes. JCI Insight 2018, 3, e98607.
- Carrasco, A.; Recabarren, M.P.; Rojas-García, P.P.; Gutiérrez, M.; Morales, K.; Sir-Petermann, T.; Recabarren, S.E. Prenatal Testosterone Exposure Disrupts Insulin Secretion and Promotes Insulin Resistance. Sci. Rep. 2020, 10, 404.
- Risal, S.; Pei, Y.; Lu, H.; Manti, M.; Fornes, R.; Pui, H.P.; Zhao, Z.; Massart, J.; Ohlsson, C.; Lindgren, E.; et al. Prenatal Androgen Exposure and Transgenerational Susceptibility to Polycystic Ovary Syndrome. Nat. Med. 2019, 25, 1894–1904.
- Ferreira, S.R.; Goyeneche, A.A.; Heber, M.F.; Abruzzese, G.A.; Ferrer, M.J.; Telleria, C.M.; Motta, A.B. Prenatal Testosterone Exposure Induces Insulin Resistance, Uterine Oxidative Stress and pro-Inflammatory Status in Rats. Mol. Cell Endocrinol. 2021, 519, 111045.
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297.
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal MiRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.
- Yu, Y.; Du, H.; Wei, S.; Feng, L.; Li, J.; Yao, F.; Zhang, M.; Hatch, G.M.; Chen, L. Adipocyte-Derived Exosomal MiR-27a Induces Insulin Resistance in Skeletal Muscle Through Repression of PPARγ. Theranostics 2018, 8, 2171–2188.
- Chen, T.; Zhang, Y.; Liu, Y.; Zhu, D.; Yu, J.; Li, G.; Sun, Z.; Wang, W.; Jiang, H.; Hong, Z. MiR-27a Promotes Insulin Resistance and Mediates Glucose Metabolism by Targeting PPAR-γ-Mediated PI3K/AKT Signaling. Aging 2019, 11, 7510–7524.
- Jiang, X.; Li, J.; Zhang, B.; Hu, J.; Ma, J.; Cui, L.; Chen, Z.J. Differential Expression Profile of Plasma Exosomal MicroRNAs in Women with Polycystic Ovary Syndrome. Fertil. Steril. 2021, 115, 782–792.
- Moghetti, P.; Tosi, F. Insulin Resistance and PCOS: Chicken or Egg? J. Endocrinol. Investig. 2021, 44, 233–244.
- Nestler, J.E.; Jakubowicz, D.J. Decreases in Ovarian Cytochrome P450c17 Alpha Activity and Serum Free Testosterone after Reduction of Insulin Secretion in Polycystic Ovary Syndrome. N. Engl. J. Med. 1996, 335, 617–623.
- Cadagan, D.; Khan, R.; Amer, S. Thecal Cell Sensitivity to Luteinizing Hormone and Insulin in Polycystic Ovarian Syndrome. Reprod. Biol. 2016, 16, 53–60.
- Kim, H.H.; DiVall, S.A.; Deneau, R.M.; Wolfe, A. Insulin Regulation of GnRH Gene Expression through MAP Kinase Signaling Pathways. Mol. Cell. Endocrinol. 2005, 242, 42–49.
- Kinyua, A.W.; Doan, K.V.; Yang, D.J.; Huynh, M.K.Q.; Choi, Y.H.; Shin, D.M.; Kim, K.W. Insulin Regulates Adrenal Steroidogenesis by Stabilizing SF-1 Activity. Sci. Rep. 2018, 8, 5025.
- Deswal, R.; Yadav, A.; Dang, A.S. Sex Hormone Binding Globulin—An Important Biomarker for Predicting PCOS Risk: A Systematic Review and Meta-Analysis. Syst. Biol. Reprod. Med. 2018, 64, 12–24.
More
Information
Subjects:
Obstetrics & Gynaecology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
21 Apr 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No