Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pan Pantziarka | -- | 2002 | 2022-04-20 12:31:40 | | | |
| 2 | Dean Liu | -2 word(s) | 2000 | 2022-04-21 04:09:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pantziarka, P.; , .; Blagden, S. Inhibiting the Priming for Cancer in Li-Fraumeni Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/22007 (accessed on 04 July 2026).
Pantziarka P, , Blagden S. Inhibiting the Priming for Cancer in Li-Fraumeni Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/22007. Accessed July 04, 2026.
Pantziarka, Pan, , Sarah Blagden. "Inhibiting the Priming for Cancer in Li-Fraumeni Syndrome" Encyclopedia, https://encyclopedia.pub/entry/22007 (accessed July 04, 2026).
Pantziarka, P., , ., & Blagden, S. (2022, April 20). Inhibiting the Priming for Cancer in Li-Fraumeni Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/22007
Pantziarka, Pan, et al. "Inhibiting the Priming for Cancer in Li-Fraumeni Syndrome." Encyclopedia. Web. 20 April, 2022.
Copy Citation
Li-Fraumeni Syndrome (LFS) is a rare cancer pre-disposition syndrome associated with a germline mutation in the TP53 tumour suppressor gene. People with LFS have a 90% chance of suffering one or more cancers in their lifetime. No treatments exist to reduce this cancer risk.
Li-Fraumeni Syndrome
TP53
drug re-purposing
pre-cancer niche
1. Introduction
Li-Fraumeni Syndrome (LFS) is a rare autosomal dominant genetic condition that pre-disposes sufferers to develop one or more cancers [1][2]. It is associated with pathogenic germ-line variants in TP53 and has an estimated penetrance of 80–90%, higher than in other cancer pre-disposition syndromes, for example that associated with BRCA1 or BRCA2 [2][3][4]. LFS is associated with a range of cancers including bone and soft tissue sarcomas, early onset breast cancer, choroid plexus carcinoma and adrenocortical carcinoma [1][4]. Although these are considered the ‘signature’ cancers associated with a germline TP53 mutation, it should be noted that LFS pre-disposes one to a far wider range of cancers, including leukaemia, lung cancer and other more common malignancies. In addition to an unusual range of cancer types, there also appears to be a temporal component to cancer incidence, with peaks in childhood and again in the early-30s, the latter associated with the high rate of breast cancer in women [4]. The risk of developing subsequent primary cancers after the first are also very high, with data showing that 49% of people with LFS go on to develop one or more primaries within a median of 10 years [5].
While most people with LFS have inherited a pathogenic TP53 variant, there are also de novo cases, with estimates ranging up to 20% of diagnosed cases [6]. However, such estimates may be subject to detection bias as lower penetrant de novo variants are detected at a lower rate than highly penetrant variants and inherited cases.
2. The Pre-Cancer Niche
The concept of the pre-metastatic niche, proposed by David Lyden and colleagues, posits that the process of metastasis depends as much on the preparation of the receptive and supportive micro-environment at the metastatic site as it does on the properties of the metastatic cell itself [7][8]. Crucially, the process depends on factors secreted by the primary tumour to prime the niche sites to create a permissive environment for the growth of circulating tumour cells which can preferentially home to the niche and take root there. An extension of this idea describes the evolution of the primary cancer niche as a multi-step process of carcinogenesis [9].
The pre-cancerous niche hypothesis proposes that a similar process precedes the establishment of primary tumours in LFS [10]. In particular, the hypothesis suggests that pathogenic germline TP53 variants facilitate the creation of these pre-malignant niches. Key drivers for this process are: chronic inflammation and oxidative stress; pro-angiogenic signalling; immune dysregulation; metabolic plasticity; and tissue-specific interactions—with aberrant p53 as a central driver of the process. Furthermore, the pre-cancerous niche itself may cause additional mutational events to occur in local cells which, together with a defective apoptotic apparatus, ensures that cancer initiation takes place in an environment that is permissive and supportive of transformed cells.
Certain phenotypic features that are common to healthy (i.e., non-cancer-carrying), people with LFS add support to this hypothesis. The relationship between chronic inflammation, oxidative stress and p53 signalling is well characterised in numerous pre-cancerous or inflammatory conditions [11][12][13][14]. It is also known that cancer-free LFS sufferers exhibit clinical signs of increased levels of oxidative stress compared to a paired group of non-affected family members (i.e., without TP53 mutations) [15]. Furthermore, mutant p53 has been shown to fine-tune anti-oxidant responses, via NRF2, to support the survival of transformed cells [16].
A pro-angiogenic micro-environment is also a factor in the pre-cancerous niche. Fibroblasts derived from LFS patients confirm that loss of the wild-type p53 allele is sufficient to decrease TSP-1 expression and an increase in VEGF [17]. There is also some evidence that gain-of-function (GOF) TP53 mutations (including R175H and R273H common in people with LFS) have been shown to have tumour angiogenesis-promoting activity [18][19].
Metabolic plasticity is one of the hallmarks of cancer in which p53 signalling plays a central role [20]. Evidence from many cancer types, including from some ‘core’ LFS cancers such as osteosarcoma [21], show the emergence of complex metabolic pockets or compartments within tumours and stroma, including Warburg and reverse Warburg phenotypes, such that metabolites are shuttled between compartments in a process of metabolic adaptation [22][23][24]. In the case of LFS, it has been proposed that such a complex evolutionary process, driven in large part by increased oxidative stress and aberrant p53, is a major factor in carcinogenesis [25]. Notably, a key marker for this multi-compartment metabolic phenotype is loss of stromal cav-1 expression [26], and this finding has been confirmed in people with LFS compared to non-affected family members [27]. Other studies have shown that people with LFS have increased oxidative metabolism compared to non-carriers, a finding in line with data from murine models of LFS [28].
To date, there has been no published analysis examining differences in immune responses between family members harbouring pathogenic TP53 variants and related wild-type carriers. However, the role of p53 in the immune response is complex, with both direct and indirect effects reported [29]. A review summarises the data on the impact of mutant p53 on immune dysfunction, showing that it contributes to the creation of a pro-tumour micro-environment, particularly via up-regulation of NF-kB [30]. Mutant p53 also targets toll-like receptor activity in response to chronic inflammation and increased oxidative stress [31][32]. Indirect effects on immunity arise from the metabolic changes induced by mutant p53, for example via increased acidity in the micro-environment.
Given the very specific pattern of LFS cancers, and the fact that the same pathogenic variants within families may manifest in a wide range of cancer types, it is difficult to correlate specific variants with clinical variability [33]. Tissue-specific factors may play a role here. For example, a comparison of breast adipose tissue between women with and without LFS showed a statistically significantly increased aromatase expression in the LFS women [34]. Furthermore, it is also showed that prostaglandin E2 (PGE2), a key inflammatory factor, acts as a negative regulator of p53. This finding may in part explain the distinct pattern of breast cancer phenotypes in women with LFS, with data showing that 84% of invasive tumours were hormone-responsive (ER and/or PR), with a majority of these also being positive for Her2/neu, figures which are higher than for the non-LFS population [35].
Together, these various factors, summarised in Figure 1, combine to form pre-cancerous niches which can both drive malignant transformation in individual cells and provide a supportive environment for these transformed cells to proliferate and ultimately form tumours [10]. Again, evidence from non-cancer-bearing individuals with LFS supports such a hypothesis. A comparison of telomere length of people with LFS compared to non-affected family members shows that TP53 mutation carriers have shorter telomeres [36][37], which may be related to the age of cancer onset [38][39]. Researchers posit that telomere attrition, a process that is exacerbated by the oxidative and other cellular stresses previously described, eventually leads to telomere crisis and consequent DNA damage and, hence, malignant transformation, as shown in Figure 2. Alternatively, the high levels of oxidative stress cause additional DNA damage, including loss of heterozygosity, and initiate malignant transformation. There is supporting evidence that cells from LFS patients display greater levels of DNA damage (chromosomal instability, senescence, etc.) [40][41][42]. One could characterise this process of accelerated telomere attrition and/or oxidative damage due to pathogenic TP53 variants as a form of accelerated host aging—again, this is a known factor in carcinogenesis [43]. DNA methylation age (Horvath age) is an epigenetic measure of aging that correlates with telomere length and can provide an alternative measure of accelerated aging [44]. Recent data show that Horvath age differed from chronological age in people with LFS compared to non-LFS individuals and that the accelerated aging was associated with cancer incidence [45]. It should also be noted that maternal stress is associated with shorter telomere length in children [46]. Given the severe psychological stress associated with LFS and incidences of cancer, it may be that this is a factor in explaining the younger age of cancer onset in succeeding generations in LFS families (genetic anticipation) [39][47].

Figure 1. Mutated p53 function leads to the creation of a pre-cancerous niche. Mutant p53 drives the creation of specific biological niches in which chronic inflammatory responses, including elevated basal oxidative stress, metabolic reprogramming, the release of pro-angiogenic factors and immune dysregulation combine to create the conditions conducive to malignant transformation. Increased oxidative stress and/or telomere attrition and tissue-specific factors, for example increased aromatase expression in the breast, contribute to additional genetic events leading to cancer initiation that arises in these cancer-supporting niches.
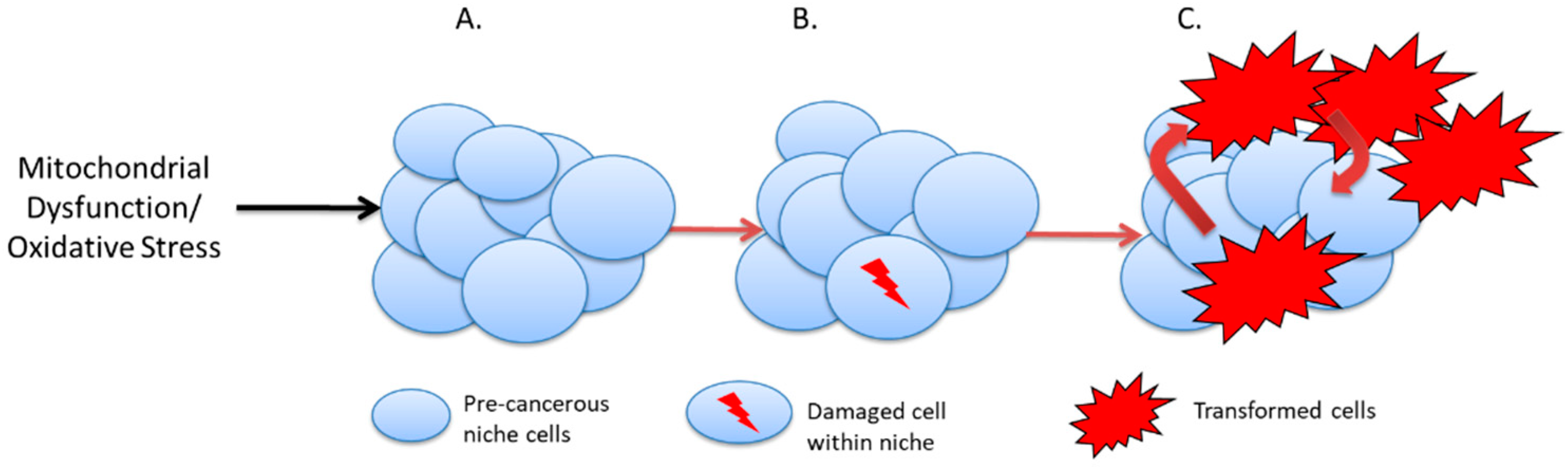
Figure 2. Cancer initiation in the pre-cancer niche. (A). Cells within the precancerous niche undergo telomere attrition or suffer genetic damage due to elevated oxidative stress. (B). Telomere crisis or further DNA damage may lead to loss of heterozygosity and malignant transformation. (C). Malignant cells in contact with chronically inflamed pre-cancerous niches proliferate and initiate tumour growth.
An interesting experimental illustration of this process comes from an animal model of LFS in which mice differing in Trp53 status were treated with either surgical implantation of a foreign object to induce chronic inflammation or a sham operation [48]. In 30/38 (79%) cases, mice with heterozygous Trp53 developed sarcomas around the implant site at a mean of 46 weeks, compared to one (10%) of the wild-type mice at 56 weeks. No sarcomas developed at the sites of sham operation, and 2/10 (20%) control heterozygous mice (with no implant) also developed sarcomas at a mean age of 80 weeks. Significantly, 90% of implant-induced sarcomas showed loss of heterozygosity, suggesting a causative effect from the chronic inflammation induced by the implant.
One obvious consequence of this view of carcinogenesis in LFS is that altering or ameliorating the pro-tumour elements of the pre-cancer niche may therefore reduce the incidence of cancer.
3. Drugging the Undruggable
Lynch syndrome, also known as hereditary non-polyposis colorectal cancer (HNPCC), is another autosomal dominant cancer pre-disposition syndrome that is associated with elevated risks of colorectal, endometrial and other cancers [49][50]. Long-term follow-up of the CAPP2 double-blind randomised placebo-controlled trial of aspirin in people with Lynch syndrome showed that it reduced the incidence of colorectal cancer, with a significantly reduced hazard ratio (HR) of 0.65 (95% CI 0.43–0.97; p = 0.035) for aspirin versus placebo, although no effect was shown in non-colorectal cancers [51].
These results are significant not only for patients with Lynch syndrome and clinicians but also for other cancer pre-disposition syndromes. It illustrates the case that the cancer risks arising from genetic pre-disposition can be reduced using medications that impact the pathways associated with carcinogenesis. Furthermore, the efficacy of the drug intervention can be shown through the use of appropriate clinical trial designs.
The specific use of aspirin is further interesting in that it may be paradigmatic of the broader approach to reducing cancer incidence in very high-risk populations. Notably, the approach has been to use an existing, licensed drug rather than to create a new molecular entity—in other words, a development approach based on drug re-purposing. Re-purposing benefits from the use of existing data on drug safety, posology, pharmacokinetics and knowledge of mechanisms of action, in addition to the easy availability of the medications and lower drug costs, particularly in the case of generics [52][53]. In the case of cancer prevention in specific high-risk populations, it is likely that treatments will extend for many years, perhaps even across a patient’s lifetime; therefore, candidate drugs must have long-term safety and tolerability data. Drugs used for chronic diseases or designed for long-term use may therefore have additional benefit as re-purposing candidates. In contrast, there can be no long-term data on newly developed medicines. One consequence is that clinical trials investigating the cancer prevention effects of re-purposed drugs can proceed relatively quickly, as early phase safety trials are not required if the drug is going to be used at a similar dose and schedule to the original use of the drug. It is also worth noting that when treatment extends for many years, drug costs will also become a factor in health technology assessment; therefore, low-cost re-purposing candidates may also be more attractive in this respect.
References
- Malkin, D. Li-fraumeni syndrome. Genes Cancer 2011, 2, 475–484.
- Schneider, K.; Zelley, K.; Nichols, K.E.; Garber, J. Li-Fraumeni Syndrome; University of Washington: Seattle, WA, USA, 2021.
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238.
- Amadou, A.; Waddington Achatz, M.I.; Hainaut, P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: Temporal phases of Li-Fraumeni syndrome. Curr. Opin. Oncol. 2018, 30, 23–29.
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681.
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum. Mutat. 2014, 35, 654–662.
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “soil”: The premetastatic niche. Cancer Res. 2006, 66, 11089–11093.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518.
- Pantziarka, P. Primed for cancer: Li Fraumeni Syndrome and the pre-cancerous niche. Ecancermedicalscience 2015, 9, 541.
- Staib, F.; Robles, A.I.; Varticovski, L.; Wang, X.W.; Zeeberg, B.R.; Sirotin, M.; Zhurkin, V.B.; Hofseth, L.J.; Hussain, S.P.; Weinstein, J.N.; et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005, 65, 10255–10264.
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337.
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646.
- Yamanishi, Y.; Boyle, D.L.; Pinkoski, M.J.; Mahboubi, A.; Lin, T.; Han, Z.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 2002, 160, 123–130.
- Macedo, G.S.; Lisbôa da Motta, L.; Giacomazzi, J.; Netto, C.B.O.; Manfredini, V.; Vanzin, C.S.; Vargas, C.R.; Hainaut, P.; Klamt, F.; Ashton-Prolla, P. Increased Oxidative Damage in Carriers of the Germline TP53 p.R337H Mutation. PLoS ONE 2012, 7, e47010.
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523.
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994, 265, 1582–1584.
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093.
- Xu, J.; Qian, J.; Hu, Y.; Wang, J.; Zhou, X.; Chen, H.; Fang, J.-Y. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci. Rep. 2014, 4, 4223.
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292.
- Sotgia, F.; Martinez-Outschoorn, U.E.; Lisanti, M.P. The reverse warburg effect in osteosarcoma. Oncotarget 2014, 5, 7982–7983.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051.
- Bonuccelli, G.; Whitaker-Menezes, D.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Witkiewicz, A.K.; Vander Heiden, M.G.; Migneco, G.; Chiavarina, B.; et al. The reverse Warburg effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010, 9, 1960–1971.
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res. 2012, 72, 5130–5140.
- Pantziarka, P. Li Fraumeni syndrome, cancer and senescence: A new hypothesis. Cancer Cell Int. 2013, 13, 35.
- Mercier, I.; Camacho, J.; Titchen, K.; Gonzales, D.M.; Quann, K.; Bryant, K.G.; Molchansky, A.; Milliman, J.N.; Whitaker-Menezes, D.; Sotgia, F.; et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: Chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am. J. Pathol. 2012, 181, 278–293.
- Sherif, Z.A.; Sultan, A.S. Divergent control of Cav-1 expression in non-cancerous Li-Fraumeni syndrome and human cancer cell lines. Cancer Biol. Ther. 2013, 14, 29–38.
- Wang, P.-Y.; Ma, W.; Park, J.-Y.; Celi, F.S.; Arena, R.; Choi, J.W.; Ali, Q.A.; Tripodi, D.J.; Zhuang, J.; Lago, C.U.; et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med. 2013, 368, 1027–1032.
- Shi, D.; Jiang, P. A Different Facet of p53 Function: Regulation of Immunity and Inflammation During Tumor Development. Front. Cell Dev. Biol. 2021, 9, 762651.
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2021, 8, 1868.
- Shatz, M.; Menendez, D.; Resnick, M.A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012, 72, 3948–3957.
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011, 7, e1001360.
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li-Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988.
- Wang, X.; Docanto, M.M.; Sasano, H.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer; Lo, C.; Simpson, E.R.; Brown, K.A. Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: A novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Res. 2015, 75, 645–655.
- Masciari, S.; Dillon, D.A.; Rath, M.; Robson, M.; Weitzel, J.N.; Balmana, J.; Gruber, S.B.; Ford, J.M.; Euhus, D.; Lebensohn, A.; et al. Breast cancer phenotype in women with TP53 germline mutations: A Li-Fraumeni syndrome consortium effort. Breast Cancer Res. Treat. 2012, 133, 1125–1130.
- Trkova, M.; Prochazkova, K.; Krutilkova, V.; Sumerauer, D.; Sedlacek, Z. Telomere length in peripheral blood cells of germline TP53 mutation carriers is shorter than that of normal individuals of corresponding age. Cancer 2007, 110, 694–702.
- Kruk, P.A.; Bohr, V.A. Telomeric length in individuals and cell lines with altered p53 status. Radiat. Oncol. Investig. 1999, 7, 13–21.
- Pinto, C.; Veiga, I.; Pinheiro, M.; Peixoto, A.; Pinto, A.; Lopes, J.M.; Reis, R.M.; Oliveira, C.; Baptista, M.; Roque, L.; et al. TP53 germline mutations in Portugal and genetic modifiers of age at cancer onset. Fam. Cancer 2009, 8, 383–390.
- Tabori, U.; Nanda, S.; Druker, H.; Lees, J.; Malkin, D. Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome. Cancer Res. 2007, 67, 1415–1418.
- Boyle, J.M.; Mitchell, E.L.; Greaves, M.J.; Roberts, S.A.; Tricker, K.; Burt, E.; Varley, J.M.; Birch, J.M.; Scott, D. Chromosome instability is a predominant trait of fibroblasts from Li-Fraumeni families. Br. J. Cancer 1998, 77, 2181–2192.
- Bischoff, F.Z.; Yim, S.O.; Pathak, S.; Grant, G.; Siciliano, M.J.; Giovanella, B.C.; Strong, L.C.; Tainsky, M.A. Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: Aneuploidy and immortalization. Cancer Res. 1990, 50, 7979–7984.
- Shay, J.W.; Tomlinson, G.; Piatyszek, M.A.; Gollahon, L.S. Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol. Cell. Biol. 1995, 15, 425–432.
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Pestell, R.G.; Howell, A.; Sotgia, F. Accelerated aging in the tumor microenvironment: Connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle 2011, 10, 2059–2063.
- Pearce, E.E.; Horvath, S.; Katta, S.; Dagnall, C.; Aubert, G.; Hicks, B.D.; Spellman, S.R.; Katki, H.; Savage, S.A.; Alsaggaf, R.; et al. DNA-methylation-based telomere length estimator: Comparisons with measurements from flow FISH and qPCR. Aging 2021, 13, 14675–14686.
- Pienkowska, M.; Samuel, N.; Choufani, S.; Subasri, V.; Patel, N.; Weksberg, R.; Kafri, R.; Malkin, D. Abstract 2114: Horvath clock as a predictor of cancer risk in LFS patients. Cancer Res. 2021, 81, 2114.
- Stout-Oswald, S.A.; Glynn, L.M.; Bisoffi, M.; Demers, C.H.; Davis, E.P. Prenatal exposure to maternal psychological distress and telomere length in childhood. Dev. Psychobiol. 2022, 64, e22238.
- Brown, B.W.; Costello, T.J.; Hwang, S.-J.; Strong, L.C. Generation or birth cohort effect on cancer risk in Li-Fraumeni syndrome. Hum. Genet. 2005, 118, 489–498.
- Tazawa, H.; Tatemichi, M.; Sawa, T.; Gilibert, I.; Ma, N.; Hiraku, Y.; Donehower, L.A.; Ohgaki, H.; Kawanishi, S.; Ohshima, H. Oxidative and nitrative stress caused by subcutaneous implantation of a foreign body accelerates sarcoma development in Trp53+/− mice. Carcinogenesis 2007, 28, 191–198.
- Bhattacharya, P.; McHugh, T.W. Lynch Syndrome; StatPearls: Treasure Island, FL, USA, 2021.
- Li, X.; Liu, G.; Wu, W. Recent advances in Lynch syndrome. Exp. Hematol. Oncol. 2021, 10, 37.
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863.
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58.
- Pantziarka, P.; Verbaanderd, C.; Huys, I.; Bouche, G.; Meheus, L. Repurposing drugs in oncology: From candidate selection to clinical adoption. Semin. Cancer Biol. 2021, 68, 186–191.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
720
Revisions:
2 times
(View History)
Update Date:
21 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No