Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Mubarak | -- | 2775 | 2022-04-18 18:27:34 | | | |
| 2 | Conner Chen | Meta information modification | 2775 | 2022-04-19 02:40:20 | | | | |
| 3 | Conner Chen | Meta information modification | 2775 | 2022-04-22 03:50:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mubarak, M.; Mechchate, H.; , .; El Omari, N.; El Hachlafi, N.; Shariati, M.A.; Wilairatana, P.; Bouyahya, A. Aging and Its Molecular Mechanism. Encyclopedia. Available online: https://encyclopedia.pub/entry/21899 (accessed on 06 August 2026).
Mubarak M, Mechchate H, , El Omari N, El Hachlafi N, Shariati MA, et al. Aging and Its Molecular Mechanism. Encyclopedia. Available at: https://encyclopedia.pub/entry/21899. Accessed August 06, 2026.
Mubarak, Mohammad, Hamza Mechchate, , Nasreddine El Omari, Naoufal El Hachlafi, Mohammad Ali Shariati, Polrat Wilairatana, Abdelhakim Bouyahya. "Aging and Its Molecular Mechanism" Encyclopedia, https://encyclopedia.pub/entry/21899 (accessed August 06, 2026).
Mubarak, M., Mechchate, H., , ., El Omari, N., El Hachlafi, N., Shariati, M.A., Wilairatana, P., & Bouyahya, A. (2022, April 18). Aging and Its Molecular Mechanism. In Encyclopedia. https://encyclopedia.pub/entry/21899
Mubarak, Mohammad, et al. "Aging and Its Molecular Mechanism." Encyclopedia. Web. 18 April, 2022.
Copy Citation
Aging is a continuous process over time that is mainly related to natural alterations in mechanical–biological processes. This phenomenon is due to several factors, including the time and energy of biological processes. Aging can be attributed to biological factors such as oxidative stress, cell longevity, and stem cell senescence.
aging
senescence
1. Introduction
Higher organisms are organized into organs and tissues with complex interconnections. During the life of a higher organism, the loss of functional and structural cells is directly compensated for by stem cells. However, over time, the ability of these stem cells to generate newly differentiated cells weakens for two reasons: the depletion of stem cells and the reduction in the energy necessary for the survival of these cells (ΔG), which is lost in the form of ΔS [1][2][3][4]. This mechanical–biological process results in aging, which is essentially associated with the senescence of the cells constituting the organism (their inability to divide and the inability of cells to be renewed). Thus, this process is becoming more important with the increase in life expectancy leading to the appearance of a certain number of so-called age-related diseases. Several recent works have deciphered the molecular nature of certain pathologies. They have demonstrated a close relationship between the aging process and several human pathologies, particularly cancer, Alzheimer’s, and Parkinson’s diseases [5][6][7].
2. Aging and Its Molecular Mechanism
Human aging, unlike sickness, is a progressive time-related process. It varies from person to person, and corresponds biologically to a loss of homeostasis, an increase in the organism’s sensitivity and susceptibility to disease and death, and the progressive degeneration of cells, tissues, and organs associated with advancing age [8]. These aspects of deterioration are called senescence, and are responsible for the weakening of an individual’s health. They also cause physiological changes in ‘‘regular’’ aging, such as menopause and decreased kidney function, and age-related disorders, such as coronary heart diseases, in ‘‘ordinary’’ aging. Several variables contribute to the loss of homeostasis, which is ultimately the consequence of a genetic program; some models propose that genes function to increase or decrease the relative risk of death by increasing the likelihood of disease [8]. Changes in the crystal structure or macromolecular aggregation at the molecular level, the loss and shortening of telomeres at the chromosomal level, changes in mitochondria, and the accumulation of lipofuscins inducing cellular aging are all monitored as part of the aging process [8], along with the appearance of cross-linking lesions of collagen and elastic fibers, and amyloid deposition. This usually causes a change in an organism’s appearance, function, and behavior [8][9].
The human average life duration has risen substantially over time; the most remarkable possible lifespan has remained stable, ranging from 90 to 100 years, and differs from person to person. The average human life expectancy has increased in recent years due to changes and developments in disease management and the socio-economic status and nutritional status of individuals, as well as fewer accidents; the improvement in all these factors has contributed to an increase in the average human life expectancy [10]. At the very least, increased mortality after maturation [11][12][13], changes in the biochemical composition of tissues [14], progressive declines in physiological capacity [15][16][17], and reduced abilities to respond adaptively to environmental stimuli are among the numerous patterns associated with aging in mammals. However, these are as yet unclear and unverified, because the mechanisms of aging could be highly different between animals, tissues, and cells.
Aging mechanisms in humans differ significantly from those of one organism, tissue, or cell, making it impossible to establish a specific mechanism. On the other hand, other academics have focused on understanding the evolutionary foundation of senescence in the aging mechanism so as to understand the aging process and study the various genes involved in senescence, which can be divided into three groups; namely, genes regulating somatic maintenance and repair, genes promoting early survival, and genes causing late deleterious mutations [18][19]. These essential genes have been shown to impact the evolution and survival of a species, and may play a role in reducing longevity due to higher growth and repeatability. The published research in molecular genetics has revealed that cellular senescence could have an opposite pleiotropic effect, preventing cancer while simultaneously contributing to organism aging. Figure 1 describes the molecular mechanisms involved in the aging process.
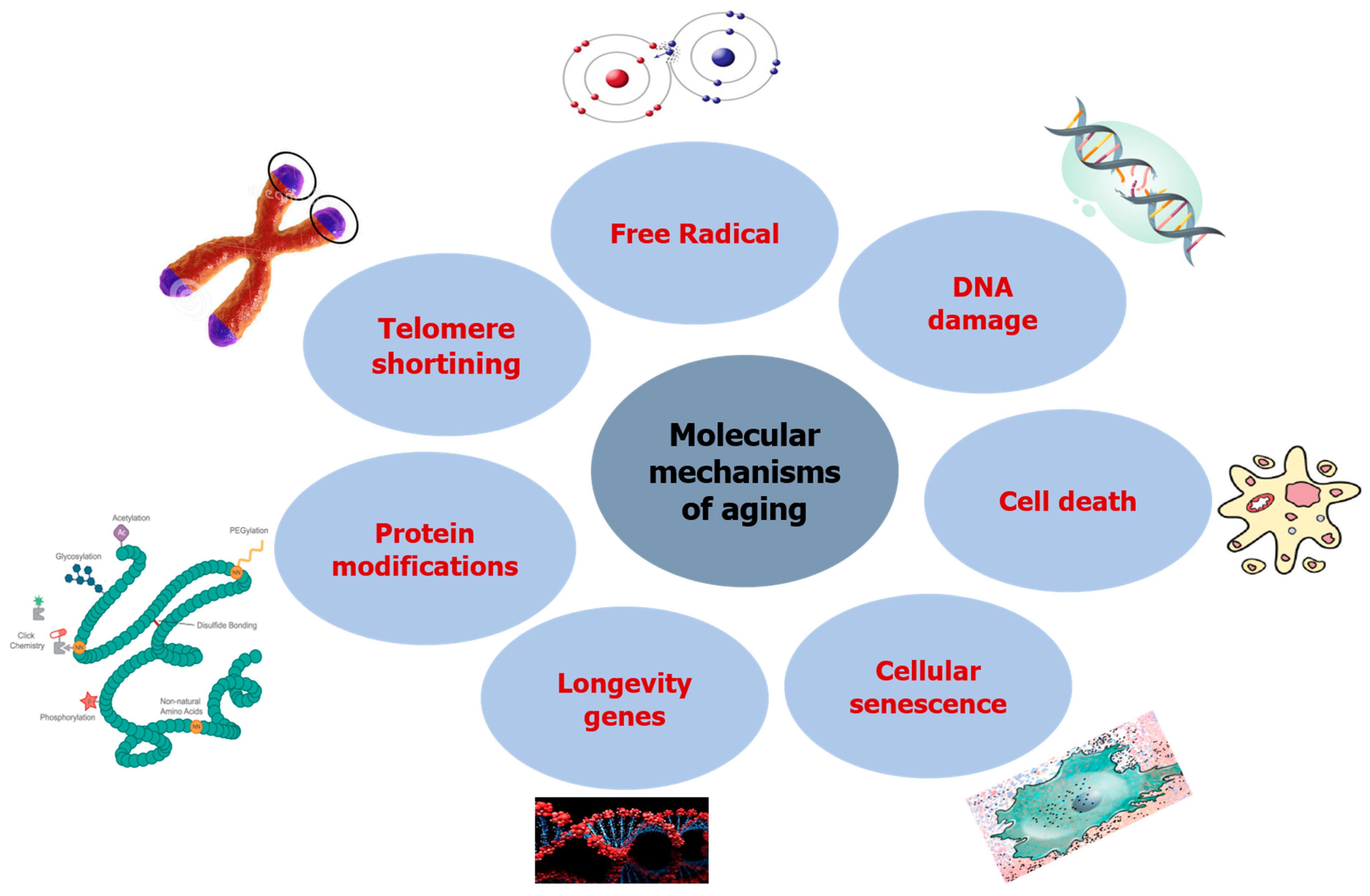
Figure 1. Molecular mechanisms inducing aging.
2.1. DNA Damage
The molecular mechanisms of aging are still unknown. According to researchers, aging is simply a physiological decline caused by the accumulation of random damage to vital molecules in aging populations. It is an example of background radiation causing genetic damage that leads to mutations, resulting in functional impairment and death [20][21]. The primary example of this theory is the ability to repair DNA damage caused by intrinsic sources such as replication defects and chemical modifications of DNA [22][23], or by external sources, including UV and genotoxic drugs [22][23] in species with different lifespans [24]. Mutations, transcription, replication halts, and the DNA damage response are triggered when DNA is damaged (DDR). These DDRs block cell cycle progression and activate signaling pathways affecting the cell via repair, apoptosis, or cellular senescence [25]. In addition, the presence of defects in genes involved in the DNA repair system causes the accumulation of unrepaired DNA and chromosomal damage [26].
On the other hand, exonuclease 1 (EXO1) and postmeiotic segregation increased 2 (PMS2) are two components that play a vital role in the DNA repair system. Their function contributes to human longevity [27][28]. Indeed, various investigations in human patients and mouse models have highlighted the relevance of DNA repair in the aging process [29]. Thus, it is not the DNA damage that causes aging; instead, it is about how the cell reacts to DNA damage and how this response will affect the organism’s life. In this respect, any abnormality or mutation in DNA repair pathways—such as Werner’s syndrome caused by a mutation in Werner’s syndrome protein (WRN), a gene that encodes a RecQ DNA helicase essential for replication stress management and telomere stability [25][30]—causes rapid aging and shorter lifespan [31]. Furthermore, researchers discovered several clinical disorders caused by problems with the genome maintenance system and aging-related syndromes. For example, Cockayne syndrome is affected by transcription-coupled nucleotide excision repair (TC-NER) [9][32]—these DNA damage repairs are generally induced after the exposure of genetic material to UV rays; ataxia-telangiectasia (AT) affected by DNA damage response—an immune deficiency affecting the humoral pathway and manifested by progressive cerebellar ataxia, linked to genetic instability of the genes that code for a protein kinase (coded by the MRE11 gene) controlling the repair of DNA double-strand breaks, especially in cerebellar and endothelial cells; Werner syndrome impacted by telomere maintenance and replication stress [8][33]—an autosomal recessive genetic disease inducing premature aging due to genetic instability, which affects the DNA repair system; Rothmund–Thomson syndrome, affected by DNA replication starting codes [34][35]—a hereditary genetic dermatosis, manifesting in dermatological signs linked to premature aging, which predisposes one to skin cancer.
2.2. Free Radicals
It has been discovered that the majority of age-related alterations are caused by molecular damage induced by free radical [36][37][38] atoms or molecules possessing an unpaired and reactive electron, constituting another possible cause of aging. These oxygen-derived species can react with macromolecules to produce free radicals from the attacked molecules [38][39], and act as secondary messengers in signaling pathways implicated in the control of various mechanisms, such as changes in gene expression, cell replication, differentiation, and apoptotic cell death [40][41][42]. The generation of these free radicals in human organs such as the heart, kidney, and liver affects maximal lifespan [43][44]. In this context, nutritional antioxidants have been found to reduce the risk of vascular dementia, heart disease, and cancer in humans [45][46]. Reactive oxygen species (ROS), in turn, play a role in the somatic accumulation of mutations in mitochondrial DNA, which is one of the developmental–genetic aspects of aging. These mutations result in a gradual loss of bioenergetic capacity, as well as aging and cell death [47][48][49]—‘‘Mitochondrial aging’s redox mechanism’’ [50]. As people age, oxygenated free radicals play a role in mitochondrial DNA (mtDNA) damage [51][52][53]. This damage causes inefficient mitochondrial respiration, which increases with age, as well as the onset of age-related illnesses, including Parkinson’s disease [54][55], Alzheimer’s disease [56][57], Huntington’s chorea [58][59], and others. mtDNA is passed down through the generations and replicates. Moreover, it is much more susceptible to mutations than nuclear DNA, and the rate of mutations rises with age [60][61].
2.3. Telomere Shortening
In addition to DNA damage, telomere length is associated with age-related disorders, whereby telomere shortening can impair somatic stem cell function. Findings indicate that telomerase-deficient mice have short telomeres and age prematurely, whereas cancer-resistant mice with high telomerase expression have long telomeres and age more slowly [62][63]. The phenomenon of telomere shortening also remains a feature and a counting mechanism of senescent cells [64][65]. Telomeres consist of long stretches of TTAGGG repeats located at the ends of chromosomes, and act as protectors that prevent them from degrading or fusing with other chromosome ends [51]. Telomerase expression is limited in human somatic cells, leading to telomere reduction and replicative senescence [52]. In fibroblasts and peripheral blood lymphocytes, the average length of the terminal restriction fragment of chromosomes decreases with age [66][67][68]. These findings show that telomere length, rather than telomerase activity, is the most critical determinant in cellular aging. Furthermore, the shortest telomere affects cell viability and chromosome stability, rather than the average-length telomeres. In this respect, several studies have demonstrated that in aged animals and humans, telomeres shorten over time [62][69]. Shorter telomeres are associated with an increased risk of death [70] and replicative senescence; they prevent cancer cells from dividing indefinitely [71]. In contrast, non-enzymatic telomere elongation extends cell lifetime in the laboratory [72].
2.4. Protein Modifications
Proteins are the building blocks of living creatures’ cellular and physiological functions, and their physical and chemical qualities determine their activities and functions. Protein folding and final conformation, and biochemical activity, stability, and half-life are all affected by the primary sequence [73]. Researchers have shown that protein repair and modification might play a role in longevity in certain situations [74]. The oxidation of amino acid residues, metal-catalyzed oxidation, and change caused by lipid oxidation products reduce the specific activity of numerous enzymes, affect thermal stability, and increase the carbonyl content of proteins [75][76]. Moreover, protein acetylation has also been proposed to play a significant role in the aging process by improving the function of specific genes, most notably the AMP-activated protein kinase (AMPK) regulatory subunit, which has been linked to increased longevity [77].
2.5. Longevity Genes
Scientists propose that the aging process is primarily caused by a genetically programmed continuum of growth and maturation. The maximum lifespan is very species-specific, as humans have a maximum lifespan 30 times longer than mice. In this context, it was discovered that specific genes found in many animals play a role in determining the full lifetime potential [78][79][80]. Indeed, the existence of these genes results in the synthesis of products that are engaged in the control of the species’ life via several mechanisms, including the modulation of stress and resistance, the increase in metabolic capacity, and the silencing of genes that promote aging [81][82][83]. In this respect, numerous investigations have suggested that the overexpression of the SIR2 gene and its homolog increases the lifespan of yeasts and nematodes [84].
Sir2 has been linked to reduced histone acetylation at the amino group of N-terminal lysine residues and global hypoacetylation in yeast [85]. Sirtuins are thought to play an essential role in cell response to several stimuli, including oxidative and genotoxic stress, and are necessary for cell metabolism [86]. Many studies have questioned the direct involvement of sirtuins in extending human lifespan, and their intervention in many human body systems such as the liver and cardiovascular system. It has been suggested that the main activity of sirtuins is the deacetylation of lysine residues [87][88]. Sirtuins cleave nicotinamide adenine dinucleotide (NAD) to nicotinamide. Then, an acetyl/acyl group is transferred from the substrate to the ADP-ribose moiety of NAD, resulting in 2′-O-acetyl-ADP-ribose and a deacetylated substrate [89].
Other researchers suggest that mutations in specific genes, such as the daf-16 gene implicated in various signal transduction pathways, including insulin signaling [84][90][91], may cause greater longevity in mutants [92]. In contrast, genetic research on mammalian longevity has revealed the presence of immunological loci in mice and humans, with these loci having implications for longevity [71]. The role of genetics in longevity was highlighted after scientists discovered that siblings and parents of long-lived people also live longer, and researchers found the presence of multiple genes on chromosome 4 linked to exceptional longevity [93][94][95], most of which are pleiotropy genes. However, since age causes differences in gene expression in muscles and the brain, several studies have shown that caloric restriction prevents age-related gene expression changes in mice. Furthermore, several investigations have focused on the cellular pathway by suggesting that aging is a cellular model, and that an individual capacity is relatively proportional to the functional capacity of the cells [96][97].
Intracellular enzymes such as collagenases, elastases, and tyrosinase are increased by intrinsic aging and photoaging factors, resulting in skin aging [98]. Extrinsic aging is generated by external stimuli, such as chronic exposure to pollutants or UV rays. At the same time, it is believed that internal aging is controlled and established by several hereditary genes [99][100]. Collagen and elastase in the dermis denature as a result of persistent UV exposure and other external factors, leading to wrinkles and the photoaging of the skin; by stimulating intracellular signal transcription pathways such as p38 mitogen-activated protein kinase and c-Jun-N-terminal kinase, this mechanism will induce the creation of MMPs, which can arise from extracellular matrix (ECM) degradation [101][102][103]. Numerous proteins present inside the ECM have been discovered. Elastase is a member of the chymotrypsin family of proteases that is primarily responsible for the degradation of elastin and collagen, essential biomolecules protecting the skin against damages [104]. Several studies have emphasized the critical role of disposing foreign proteins within the ECM during neutrophil phagocytosis and tissue healing in standard settings [98]. Many research works have focused on the direct impact of inhibiting those enzymes. It has been found that inhibiting elastase and MMP-1 enzymes may positively affect skin aging due to their usefulness in avoiding skin sagging and losses of elasticity [98][105]. Furthermore, scientists have investigated the impact of inhibiting tyrosinase, a copper-containing monooxygenase that catalyzes the O-hydroxylation of tyrosine to 3,4-dihydroxyphenylalanine and then to dopaquinone, which is synthesized by epithelial, mucosal, retinal, and ciliary body melanocytes, and which is deeply involved in the protection of the skin from melanogenesis [106][107].
2.6. Cellular Senescence
Cellular senescence and the number of divisions are required to determine proliferative lifespan [1][108]. Several studies have shown that a decrease in the rate of cell proliferation corresponds to aging in animals [19][109]. Multiple intrinsic and extrinsic factors, including oxidative stress (OS), DNA damage, oncogene activation, epigenetic stress, and mitotic spindle stress, can cause cellular senescence. When stem cells or progenitor cells are damaged, cellular senescence compromises tissue function and reduces tissue regeneration capacity [110]. In this context, several studies on the senescence of many cells and their relationship with aging, such as glial cells [111], keratinocytes [112], vascular smooth muscle cells [113], lens cells [114], endothelial cells [115], and lymphocytes [116], have been conducted. The results indicate that eliminating senescent cells in adult wild-type mice delays tumor formation; thus, eradicating senescence could help people live longer [117]. In addition, normal cells in life forms have a limited proliferative ability; cells divide less frequently in humans, and their latency before proliferation increases. Despite these investigations, there is no conclusive evidence that senescent cells grow in vivo as people age.
2.7. Cell Death
Traditionally, cell death methods have been divided into energy-dependent programmed cell death apoptosis mechanisms and necrotic cell death mechanisms. The body can preserve equilibrium in the apoptosis mechanism by committing active “suicide”. This is a preprogrammed death caused by the progressive activation of a group of cysteine proteases known as caspases. Most cells have caspases in their cytoplasm, but they are generally inactive (procaspase) [118]. Because there is no release beyond the cell, there is no inflammation. This occurs in response to environmental stimuli and is controlled by genes [119][120]. In addition to its essential role in the immune system, where up to 95% of T cells undergo cell death (presumably because they recognize self-antigens) [121], it involves the compaction and segregation of chromatin adjacent to the nuclear membrane and the condensation of the cytoplasm, eventually evolving into nuclear/cellular fragmentation. Meanwhile, apoptosis may play a role in aging and age-related disorders. Apoptosis, followed by the replacement by division of another cell, may occur if cells are unable to repair DNA damage; it is also essential for wound healing, which is often reduced as people age, often in conjunction with local inflammation [122]. The central nervous system shows the most transparent relationship between apoptosis and aging, whereby neuronal apoptosis increases with aging. Similarly, cancer rates rise with age due to decreased apoptotic defenses [123].
References
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95.
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 2018, 128, 1238–1246.
- Martin, G.M. Keynote: Mechanisms of Senescence—Complificationists versus Simplificationists. Mech. Ageing Dev. 2002, 123, 65–73.
- Pedro de Magalhães, J. From Cells to Ageing: A Review of Models and Mechanisms of Cellular Senescence and Their Impact on Human Ageing. Exp. Cell Res. 2004, 300, 1–10.
- Baima, G.; Romandini, M.; Citterio, F.; Romano, F.; Aimetti, M. Periodontitis and Accelerated Biological Aging: A Geroscience Approach. J. Dent. Res. 2022, 101, 125–132.
- Brahadeeswaran, S.; Sivagurunathan, N.; Calivarathan, L. Inflammasome Signaling in the Aging Brain and Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2022, 1–17.
- Ding, Y.-N.; Wang, H.-Y.; Chen, H.-Z.; Liu, D.-P. Targeting Senescent Cells for Vascular Aging and Related Diseases. J. Mol. Cell. Cardiol. 2022, 162, 43–52.
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; Cabo, R. Measuring Biological Aging in Humans: A Quest. Aging Cell 2020, 19, e13080.
- Robinson, A.R.; Yousefzadeh, M.J.; Rozgaja, T.A.; Wang, J.; Li, X.; Tilstra, J.S.; Feldman, C.H.; Gregg, S.Q.; Johnson, C.H.; Skoda, E.M.; et al. Spontaneous DNA Damage to the Nuclear Genome Promotes Senescence, Redox Imbalance and Aging. Redox Biol. 2018, 17, 259–273.
- Park, J.H.; Yoo, Y.; Park, Y.J. Epigenetics: Linking Nutrition to Molecular Mechanisms in Aging. Prev. Nutr. Food Sci. 2017, 22, 81.
- Gompertz, B. On the Nature of the Function Expressive of the Law of Human Mortality, and on a New Mode of Determining the Value of Life Contingencies. Philos. Trans. R. Soc. Lond. 1825, 115, 513–583.
- Ventura, S.; Peters, K.; Martin, J.; Maurer, J. Births and Deaths: United States, 1996. Mon. Vital Stat. Rep. 1997, 46, 1–40.
- Kaeberlein, M.; McVey, M.; Guarente, L. Using Yeast to Discover the Fountain of Youth. Sci. Aging Knowl. Environ. Sage Ke 2001, 2001, pe1.
- Shock, N.W. Normal Human Aging: The Baltimore Longitudinal Study of Aging; U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute on Aging, Gerontology Research Center: Baltimore, MD, USA, 1984.
- Schaie, K.W.; Hofer, S.M. Longitudinal Studies in Aging Research. In Handbook of the Psychology of Aging, 5th ed.; Academic Press: San Diego, CA, USA, 2001; pp. 53–77. ISBN 978-0-12-101262-5.
- Lakatta, E.G. Changes in Cardiovascular Function with Aging. Eur. Heart J. 1990, 11, 22–29.
- Weinstein, J.R.; Anderson, S. The Aging Kidney: Physiological Changes. Adv. Chronic Kidney Dis. 2010, 17, 302–307.
- Kirkwood, T.B.L. Human Senescence. BioEssays 1996, 18, 1009–1016.
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 483–497.
- Rozhok, A.; DeGregori, J. A Generalized Theory of Age-Dependent Carcinogenesis. eLife 2019, 8, e39950.
- Piotrowski, I.; Kulcenty, K.; Suchorska, W.M.; Skrobała, A.; Skórska, M.; Kruszyna-Mochalska, M.; Kowalik, A.; Jackowiak, W.; Malicki, J. Carcinogenesis Induced by Low-Dose Radiation. Radiol. Oncol. 2017, 51, 369–377.
- Karasik, D.; Demissie, S.; Cupples, L.A.; Kiel, D.P. Disentangling the Genetic Determinants of Human Aging: Biological Age as an Alternative to the Use of Survival Measures. J. Gerontol. Ser. A 2005, 60, 574–587.
- Fraga, M.F. Genetic and Epigenetic Regulation of Aging. Curr. Opin. Immunol. 2009, 21, 446–453.
- Gredilla, R.; Sánchez-Román, I.; Gómez, A.; López-Torres, M.; Barja, G. Mitochondrial Base Excision Repair Positively Correlates with Longevity in the Liver and Heart of Mammals. GeroScience 2020, 42, 653–665.
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322.
- Hanawalt, P.C.; Gee, P.; Ho, L.; Hsu, R.K.; Kane, C.J. Genomic Heterogeneity of DNA Repair. Role in Aging. Ann. N. Y. Acad. Sci. 1992, 663, 17–25.
- Nebel, A.; Flachsbart, F.; Till, A.; Caliebe, A.; Blanché, H.; Arlt, A.; Häsler, R.; Jacobs, G.; Kleindorp, R.; Franke, A.; et al. A Functional EXO1 Promoter Variant Is Associated with Prolonged Life Expectancy in Centenarians. Mech. Ageing Dev. 2009, 130, 691–699.
- Han, J.; Ryu, S.; Moskowitz, D.M.; Rothenberg, D.; Leahy, D.J.; Atzmon, G.; Barzilai, N.; Suh, Y. Discovery of Novel Non-Synonymous SNP Variants in 988 Candidate Genes from 6 Centenarians by Target Capture and next-Generation Sequencing. Mech. Ageing Dev. 2013, 134, 478–485.
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 Collaborate in DNA Double-Strand Break Processing. Nature 2008, 455, 770–774.
- Chen, L.; Huang, S.; Lee, L.; Davalos, A.; Schiestl, R.H.; Campisi, J.; Oshima, J. WRN, the Protein Deficient in Werner Syndrome, Plays a Critical Structural Role in Optimizing DNA Repair. Aging Cell 2003, 2, 191–199. Available online: https://onlinelibrary.wiley.com/doi/full/10.1046/j.1474-9728.2003.00052.x (accessed on 28 September 2021).
- de Boer, J. Premature Aging in Mice Deficient in DNA Repair and Transcription. Science 2002, 296, 1276–1279.
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) Study: Clinical Findings in 102 Individuals and Recommendations for Care. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 483–493.
- Kudlow, B.A.; Kennedy, B.K.; Monnat, R.J. Werner and Hutchinson-Gilford Progeria Syndromes: Mechanistic Basis of Human Progeroid Diseases. Nat. Rev. Mol. Cell Biol. 2007, 8, 394–404.
- Ghosh, A.K.; Rossi, M.L.; Singh, D.K.; Dunn, C.; Ramamoorthy, M.; Croteau, D.L.; Liu, Y.; Bohr, V.A. RECQL4, the Protein Mutated in Rothmund-Thomson Syndrome, Functions in Telomere Maintenance. J. Biol. Chem. 2012, 287, 196–209.
- Croteau, D.L.; Singh, D.K.; Hoh Ferrarelli, L.; Lu, H.; Bohr, V.A. RECQL4 in Genomic Instability and Aging. Trends Genet. TIG 2012, 28, 624–631.
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300.
- Harman, D. The Aging Process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128.
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharmacogn. Rev. 2010, 4, 118–126.
- Sohal, R.S.; Weindruch, R. Oxidative Stress, Caloric Restriction, and Aging. Science 1996, 273, 59–63. Available online: https://www.science.org/doi/abs/10.1126/science.273.5271.59 (accessed on 28 September 2021).
- Sen, C.K.; Packer, L. Antioxidant and Redox Regulation of Gene Transcription. FASEB J. 1996, 10, 709–720.
- Suzuki, Y.J. Oxidant-Mediated Protein Amino Acid Conversion. Antioxidants 2019, 8, 50.
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as Stimulators of Signal Transduction. Free Radic. Biol. Med. 1997, 22, 269–285.
- Barja, G. Free Radicals and Aging. Trends Neurosci. 2004, 27, 595–600.
- Sohal, R.S.; Svensson, I.; Sohal, B.H.; Brunk, U.T. Superoxide Anion Radical Production in Different Animal Species. Mech. Ageing Dev. 1989, 49, 129–135.
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.-C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 9, 477.
- Zainal, T.A.; Oberley, T.D.; Allison, D.B.; Szweda, L.I.; Weindruch, R. Caloric Restriction of Rhesus Monkeys Lowers Oxidative Damage in Skeletal Muscle. FASEB J. 2000, 14, 1825–1836.
- Linnane, A.W.; Zhang, C.; Baumer, A.; Nagley, P. Mitochondrial DNA Mutation and the Ageing Process: Bioenergy and Pharmacological Intervention. Mutat. Res. 1992, 275, 195–208.
- Su, T.; Turnbull, D.M.; Greaves, L.C. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes 2018, 9, 182.
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240.
- Ozawa, T. Genetic and Functional Changes in Mitochondria Associated with Aging. Physiol. Rev. 1997, 77, 425–464.
- Miquel, J. Role of Mitochondria in Cell Aging. In Molecular Basis of Aging; CRC Press: Boca Raton, FL, USA, 1995; Available online: https://www.taylorfrancis.com/chapters/edit/10.1201/9780203711309-7/role-mitochondria-cell-aging-jaime-miquel (accessed on 28 September 2021).
- Katayama, M.; Tanaka, M.; Yamamoto, H.; Ohbayashi, T.; Nimura, Y.; Ozawa, T. Deleted Mitochondrial DNA in the Skeletal Muscle of Aged Individuals. Biochem. Int. 1991, 25, 47–56.
- Mohamed, H.R.H. Alleviation of Cadmium Chloride–Induced Acute Genotoxicity, Mitochondrial DNA Disruption, and ROS Generation by Chocolate Coadministration in Mice Liver and Kidney Tissues. Biol. Trace Elem. Res. 2021.
- Schapira, A.H.V.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and Disease Specificity of NADH CoQ1 Reductase (Complex I) Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 55, 2142–2145.
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21.
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative Profiling Brain Proteomes Revealed Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Brain 2019, 12, 8.
- Hoyer, S. Senile Dementia and Alzheimer’s Disease. Brain Blood Flow and Metabolism. Prog. Neuropsychopharmacol. Biol. Psychiatry 1986, 10, 447–478.
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 59–83. ISBN 978-3-319-71779-1.
- Beal, M.F. Neurochemistry and Toxin Models in Huntington’s Disease. Curr. Opin. Neurol. 1994, 7, 542–547.
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related MtDNA Mutations. Cells 2019, 8, 608.
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The Rise and Rise of Mitochondrial DNA Mutations. Open Biol. 2020, 10, 200061.
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere Biology and Age-Related Diseases. Clin. Chem. Lab. Med. CCLM 2018, 56, 1210–1222.
- Wang, J.-Y.; Peng, S.-H.; Ning, X.-H.; Li, T.; Liu, S.-J.; Liu, J.-Y.; Hong, B.-A.; Qi, N.-N.; Peng, X.; Zhou, B.-W.; et al. Shorter Telomere Length Increases Age-Related Tumor Risks in von Hippel-Lindau Disease Patients. Cancer Med. 2017, 6, 2131–2141.
- Srinivas, N.; Rachakonda, S.; Kumar, R. Telomeres and Telomere Length: A General Overview. Cancers 2020, 12, 558.
- Harley, C.B. Telomere Loss: Mitotic Clock or Genetic Time Bomb? Mutat. Res. 1991, 256, 271–282.
- Smith, E.M.; Pendlebury, D.F.; Nandakumar, J. Structural Biology of Telomeres and Telomerase. Cell. Mol. Life Sci. 2020, 77, 61–79.
- Weng, N.P.; Levine, B.L.; June, C.H.; Hodes, R.J. Human Naive and Memory T Lymphocytes Differ in Telomeric Length and Replicative Potential. Proc. Natl. Acad. Sci. USA 1995, 92, 11091–11094.
- Vaziri, H.; Schächter, F.; Uchida, I.; Wei, L.; Zhu, X.; Effros, R.; Cohen, D.; Harley, C.B. Loss of Telomeric DNA during Aging of Normal and Trisomy 21 Human Lymphocytes. Am. J. Hum. Genet. 1993, 52, 661–667.
- Shu, Y.; Wu, M.; Yang, S.; Wang, Y.; Li, H. Association of Dietary Selenium Intake with Telomere Length in Middle-Aged and Older Adults. Clin. Nutr. 2020, 39, 3086–3091.
- Wang, Q.; Zhan, Y.; Pedersen, N.L.; Fang, F.; Hägg, S. Telomere Length and All-Cause Mortality: A Meta-Analysis. Ageing Res. Rev. 2018, 48, 11–20. Available online: https://www.sciencedirect.com/science/article/pii/S1568163718301235 (accessed on 29 September 2021).
- Hemann, M.T.; Strong, M.A.; Hao, L.-Y.; Greider, C.W. The Shortest Telomere, Not Average Telomere Length, Is Critical for Cell Viability and Chromosome Stability. Cell 2001, 107, 67–77.
- Experimental Elongation of Telomeres Extends the Lifespan of Immortal x Normal Cell Hybrids. EMBO J. 1996, 15, 1734–1741.
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein Structure Prediction from Sequence Variation. Nat. Biotechnol. 2012, 30, 1072–1080. Available online: https://www.nature.com/articles/nbt.2419 (accessed on 29 September 2021).
- Petropoulos, I.; Friguet, B. Maintenance of Proteins and Aging: The Role of Oxidized Protein Repair. Free Radic. Res. 2006, 40, 1269–1276.
- Schöneich, C. Protein Modification in Aging: An Update. Exp. Gerontol. 2006, 41, 807–812.
- Rattan, S.I.S.; Derventzi, A.; Clark, B.F.C. Protein Synthesis, Posttranslational Modifications, and Aginga. Ann. N. Y. Acad. Sci. 1992, 663, 48–62.
- Lu, J.-Y.; Lin, Y.-Y.; Zhu, H.; Chuang, L.-M.; Boeke, J.D. Protein Acetylation and Aging. Aging 2011, 3, 911–912.
- Finch, C.E.; Tanzi, R.E. Genetics of Aging. Science 1997, 278, 407–411. Available online: https://www.science.org/doi/abs/10.1126/science.278.5337.407 (accessed on 29 September 2021).
- Singh, P.P.; Demmitt, B.A.; Nath, R.D.; Brunet, A. The Genetics of Aging: A Vertebrate Perspective. Cell 2019, 177, 200–220.
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B. Alternative Splicing in Aging and Longevity. Hum. Genet. 2020, 139, 357–369.
- Murakami, S.; Johnson, T.E. A Genetic Pathway Conferring Life Extension and Resistance to UV Stress in Caenorhabditis Elegans. Genetics 1996, 143, 1207–1218.
- Kimura, K.D.; Tissenbaum, H.A.; Liu, Y.; Ruvkun, G. daf-2, an Insulin Receptor-Like Gene That Regulates Longevity and Diapause in Caenorhabditis Elegans. Science 1997, 277, 942–946.
- Zečić, A.; Braeckman, B.P. DAF-16/FoxO in Caenorhabditis Elegans and Its Role in Metabolic Remodeling. Cells 2020, 9, 109.
- Xiong, L.; Deng, N.; Zheng, B.; Li, T.; Liu, R.H. HSF-1 and SIR-2.1 Linked Insulin-like Signaling Is Involved in Goji Berry (Lycium Spp.) Extracts Promoting Lifespan Extension of Caenorhabditis Elegans. Food Funct. 2021, 12, 7851–7866.
- Jing, H.; Lin, H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375.
- Choi, J.-E.; Mostoslavsky, R. Sirtuins, Metabolism, and DNA Repair. Curr. Opin. Genet. Dev. 2014, 26, 24–32.
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 Complex and SIR2 Alone Promote Longevity in Saccharomyces Cerevisiae by Two Different Mechanisms. Genes Dev. 1999, 13, 2570–2580.
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and Their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927.
- Tong, L.; Denu, J.M. Function and Metabolism of Sirtuin Metabolite O-Acetyl-ADP-Ribose. Biochim. Biophys. Acta BBA—Proteins Proteom. 2010, 1804, 1617–1625.
- Tissenbaum, H.A. Chapter One—DAF-16: FOXO in the Context of C. Elegans. In Current Topics in Developmental Biology; Ghaffari, S., Ed.; Forkhead FOXO Transcription Factors in Development and Disease; Academic Press: Cambridge, MA, USA, 2018; Volume 127, pp. 1–21.
- Lin, K.; Dorman, J.B.; Rodan, A.; Kenyon, C. Daf-16: An HNF-3/Forkhead Family Member That Can Function to Double the Life-Span of Caenorhabditis Elegans. Science 1997, 278, 1319–1322. Available online: https://www.science.org/doi/abs/10.1126/science.278.5341.1319 (accessed on 29 September 2021).
- Lithgow, G.J.; Andersen, J.K. The Real Dorian Gray Mouse. Bioessays 2000, 22, 410–413. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/(SICI)1521-1878(200005)22:5%3C410::AID-BIES2%3E3.0.CO;2-C (accessed on 29 September 2021).
- Flurkey, K.; Papaconstantinou, J.; Harrison, D.E. The Snell Dwarf Mutation Pit1dw Can Increase Life Span in Mice. Mech. Ageing Dev. 2002, 123, 121–130.
- Perls, T.; Levenson, R.; Regan, M.; Puca, A. What Does It Take to Live to 100? Mech. Ageing Dev. 2002, 123, 231–242.
- Messaris, G.A.; Hadjinicolaou, M.; Karahalios, G.T. Why Do We Live for Much Less than 100 Years? A Fluid Mechanics View and Approach. Phys. Fluids 2017, 29, 081903. Available online: https://aip.scitation.org/doi/abs/10.1063/1.4998717 (accessed on 29 September 2021).
- Bartlett, Z. The Limited In Vitro Lifetime of Human Diploid Cell Strains; Hayflick, L., Ed.; Academic Press: Cambridge, MA, USA, 1964.
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636.
- Azmi, N.; Hashim, P.; Hashim, D.M.; Halimoon, N.; Majid, N.M.N. Anti–Elastase, Anti–Tyrosinase and Matrix Metalloproteinase–1 Inhibitory Activity of Earthworm Extracts as Potential New Anti–Aging Agent. Asian Pac. J. Trop. Biomed. 2014, 4, S348–S352.
- Tsatsou, F.; Trakatelli, M.; Patsatsi, A.; Kalokasidis, K.; Sotiriadis, D. Extrinsic Aging: UV-Mediated Skin Carcinogenesis. Dermatoendocrinology 2012, 4, 285–297.
- Vierkötter, A.; Krutmann, J. Environmental Influences on Skin Aging and Ethnic-Specific Manifestations. Dermatoendocrinology 2012, 4, 227–231.
- Zhang, S.; Duan, E. Fighting against Skin Aging: The Way from Bench to Bedside. Cell Transplant. 2018, 27, 729–738.
- Jenkins, R.R. Free Radical Chemistry. Sports Med. 1988, 5, 156–170.
- Sárdy, M. Role of Matrix Metalloproteinases in Skin Ageing. Connect. Tissue Res. 2009, 50, 132–138.
- Chapman, H.A.; Riese, R.J.; Shi, G.-P. Emerging Roles for Cysteine Proteases in Human Biology. Annu. Rev. Physiol. 1997, 59, 63–88.
- Thring, T.S.; Hili, P.; Naughton, D.P. Anti-Collagenase, Anti-Elastase and Anti-Oxidant Activities of Extracts from 21 Plants. BMC Complement. Altern. Med. 2009, 9, 27.
- Chang, T.S. Review an Update Review of Tyrosinase Inhibitors Intern. J. Mol. Sci. 2009, 10, 2440–2475.
- Wu, L.; Chen, C.; Cheng, C.; Dai, H.; Ai, Y.; Lin, C.; Chung, Y. Evaluation of Tyrosinase Inhibitory, Antioxidant, Antimicrobial, and Anti-aging Activities of Magnolia Officinalis Extracts after Aspergillus Niger Fermentation. BioMed Res. Int. 2018, 2018, 5201786.
- Sturm, G.; Cardenas, A.; Bind, M.-A.; Horvath, S.; Wang, S.; Wang, Y.; Hägg, S.; Hirano, M.; Picard, M. Human Aging DNA Methylation Signatures Are Conserved but Accelerated in Cultured Fibroblasts. Epigenetics 2019, 14, 961–976.
- Franceschi, C. Cell Proliferation, Cell Death and Aging. Aging Clin. Exp. Res. 1989, 1, 3–15.
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. Available online: https://www.annualreviews.org/doi/abs/10.1146/annurev-physiol-030212-183653 (accessed on 29 September 2021).
- Tomita, K.; Aida, J.; Izumiyama-Shimomura, N.; Nakamura, K.; Ishikawa, N.; Matsuda, Y.; Arai, T.; Ishiwata, T.; Kumasaka, T.; Takahashi-Fujigasaki, J.; et al. Changes in Telomere Length with Aging in Human Neurons and Glial Cells Revealed by Quantitative Fluorescence in Situ Hybridization Analysis. Geriatr. Gerontol. Int. 2018, 18, 1507–1512.
- Nanba, D. Human Keratinocyte Stem Cells: From Cell Biology to Cell Therapy. J. Dermatol. Sci. 2019, 96, 66–72.
- Bierman, E.L. The Effect of Donor Age on the in Vitro Life Span of Cultured Human Arterial Smooth-Muscle Cells. In Vitro 1978, 14, 951–955.
- Tassin, J.; Malaise, E.; Courtois, Y. Human Lens Cells Have an in Vitro Proliferative Capacity Inversely Proportional to the Donor Age. Exp. Cell Res. 1979, 123, 388–392.
- Mueller, S.N.; Rosen, E.M.; Levine, E.M. Cellular Senescence in a Cloned Strain of Bovine Fetal Aortic Endothelial Cells. Science 1980, 207, 889–891. Available online: https://www.science.org/doi/abs/10.1126/science.7355268 (accessed on 29 September 2021).
- Tice, R.R.; Schneider, E.L.; Kram, D.; Thorne, P. Cytokinetic Analysis of the Impaired Proliferative Response of Peripheral Lymphocytes from Aged Humans to Phytohemagglutinin. J. Exp. Med. 1979, 149, 1029–1041.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.S.; Van Deursen, J.M.; et al. Naturally Occurring P16Ink4a-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. Available online: https://www.nature.com/articles/nature16932 (accessed on 29 September 2021).
- Nguyen, T.T.M.; Gillet, G.; Popgeorgiev, N. Caspases in the Developing Central Nervous System: Apoptosis and Beyond. Front. Cell Dev. Biol. 2021, 9, 702404. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8322698/ (accessed on 29 September 2021).
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164.
- Lockshin, R.A.; Zakeri, Z. Programmed Cell Death and Apoptosis: Origins of the Theory. Nat. Rev. Mol. Cell Biol. 2001, 2, 545–550.
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758.
- Hiebert, P.R.; Granville, D.J. Granzyme B in Injury, Inflammation, and Repair. Trends Mol. Med. 2012, 18, 732–741.
- Tower, J. Programmed Cell Death in Aging. Ageing Res. Rev. 2015, 23, 90–100.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
7.0K
Revisions:
3 times
(View History)
Update Date:
22 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No