 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Domenico Mattoscio | + 6079 word(s) | 6079 | 2020-09-14 08:33:32 | | | |
| 2 | Nicole Yin | -14 word(s) | 6065 | 2020-09-28 06:21:55 | | | | |
| 3 | Nicole Yin | Meta information modification | 6065 | 2020-11-09 11:02:47 | | |
Video Upload Options
Autophagy is a catabolic pathway that accounts for degradation and recycling of cellular components to extend cell survival under stress conditions. In addition to this prominent role, recent evidence indicates that autophagy is crucially involved in the regulation of the inflammatory response, a tightly controlled process aimed at clearing the inflammatory stimulus and restoring tissue homeostasis. To be efficient and beneficial to the host, inflammation should be controlled by a resolution program, since uncontrolled inflammation is the underlying cause of many pathologies. Resolution of inflammation is an active process mediated by a variety of mediators, including the so-called specialized pro-resolving lipid mediators (SPMs), a family of endogenous lipid autacoids known to regulate leukocyte infiltration and activities, and counterbalance cytokine production. Recently, regulation of autophagic mechanisms by these mediators has emerged, uncovering unappreciated connections between inflammation resolution and autophagy. Here, we summarize mechanisms of autophagy and resolution, focusing on the contribution of autophagy in sustaining paradigmatic examples of chronic inflammatory disorders. Then, we discuss the evidence that SPMs can restore dysregulated autophagy, hypothesizing that resolution of inflammation could represent an innovative approach to modulate autophagy and its impact on the inflammatory response.
1. Introduction
Autophagy is a physiological process that regulates the degradation and recycling of waste materials and damaged intracellular components through lysosomal activities. Initially described as a catabolic pathway activated under stress conditions and aimed to extend cell survival[1], a large body of evidence indicates now that autophagy is crucial for a variety of biological processes ranging from cell metabolism, organ development, differentiation, host defense, immunity, and inflammation. As such, alteration of the autophagic pathway has been correlated with several human pathologies like viral infections, neurological disorders, senescence, cancer, and inflammatory conditions[2]. In particular, the role of autophagy as modulator of inflammation, and its involvement in chronic inflammatory diseases is now emerging[3][4][5][6]. Indeed, autophagy may act as protective mechanism in the removal of viruses and bacteria, and a failure in the clearance of pathogens sustains the persistence of inflammation. In addition, a defective autophagic pathway fails to remove unnecessary cellular components, resulting in cell dysfunction and death that act as an inflammatory trigger. Importantly, autophagy regulates the production of inflammatory mediators, thus acting as an anti-inflammatory pathway in certain conditions (reviewed in[7]), and affects the activity of key immune cells such as polymorphonuclear leukocytes (PMNs) and macrophages (MФs).
Acute inflammation is a protective response of vascularized tissues to injuries, traumas, or infections, which involves a tightly regulated and temporally restricted interaction and activation of endothelial and blood cells, as well as the production of specific chemical signals like cytokines and chemokines[8]. The physiological outcome of the inflammatory response is the restoration of tissue homeostasis and functionality, culminating in tissue repair. On the contrary, an uncontrolled inflammatory reaction can be detrimental and is indeed a driving pathogenetic mechanism for a wide range of human diseases[9]. To keep inflammation within physiologic boundaries, a proper resolution program must occur[10]. Resolution of inflammation is indeed an active process modulated by a variety of mediators, among which the so-called specialized pro-resolving lipid mediators (SPMs) play a preeminent role. These are endogenously-produced molecules that modulate leukocyte infiltration and activities, and cytokine release to terminate inflammation[11]. In addition, accumulating evidence indicates that modulation of autophagy is a relevant mechanism by which SPMs terminate the inflammatory response and restore tissue homeostasis. Failure in resolution has been associated with several diseases[12], suggesting that therapeutic induction of resolution may be proposed as an alternative approach to conventional anti-inflammatory pharmacology. Along these lines, SPMs proved beneficial in a several experimental diseases (reviewed in[13][14]) including periodontal and neurological diseases[15][16], arthritis[17], sickle cell disease[18], lung injury[19][20][21], and atherosclerosis[22] [22]. Moreover, clinical studies with human volunteers confirmed that SPMs represent safe and effective therapeutics in asthma, skin infections, and eczema[23][24][25].
Here, we initially describe key mechanisms of autophagy and resolution of inflammation, focusing on the role of autophagy in chronic inflammatory disorders. Then, we discuss the emerging functions of SPMs as modulators of the autophagic pathway, and we outline our perspectives on the interplay between pro-resolving and autophagic pathways, proposing its modulation as innovative therapeutic strategy to combat human disease.
2. The Autophagic Pathway
The term autophagy (from Greek ‘to eat one’s self’) indicates several processes whose central function is to sequester cellular materials inside double-membraned organelles and to deliver autophagic cargoes to lysosomes for degradation.
In microautophagy, the digested materials are carried to lysosomes by direct protrusion or invagination of the lysosomal membrane to directly enwrap and degrade cytosolic components[26].
In chaperone-mediated autophagy, unfolded proteins that contain a specific pentapeptide motif are translocated by the heat shock cognate (hsc) protein 70 to the lysosome for degradation[27].
In macroautophagy (simply referred as autophagy hereafter), the best studied autophagic route, a double-layered membrane named phagophore engulfs and encloses bulk cytoplasm or specific targets inside the autophagosome (Figure 1). The autophagosome then fuses with the lysosomal membrane where hydrolytic enzymes digest autophagosome cargo, and breakdown products are recycled back to cytosol for reuse[28]. Despite being initially considered as a non-selective pathway, induced as a survival mechanism in response to cellular stresses, it is now clear that autophagy is also a highly selective process involved in the clearance of protein aggregates (aggrephagy), intracellular pathogens (xenophagy), damaged or redundant organelles (such as pexophagy for peroxisome degradation, mitophagy for mithocondria, reticulophagy for endoplasmic reticulum, nucleophagy for nucleus, lysophagy for lysosomes), and macromolecular complexes (lipophagy for lipid droplets, ferritinophagy for ferritin, glycophagy for glycogen) (see[29] for a recent review on mechanisms of selective autophagy).
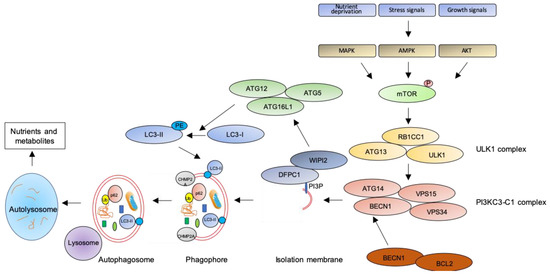
Figure 1. The autophagic pathway. See text for details.
The key step of the autophagic pathway is the formation of the autophagosome, an endomembranous organelle de novo produced in stress conditions (readers are referred to[30] for a detailed review of the molecular mechanisms of autophagosome formation). The best characterized pathway of autophagy activation is regulated by the mechanistic target of rapamycin (mTOR)[31] that senses nutrient availability. Nutrients deprivation or other environmental factors, such as oxidative stress, inhibit mTOR through AMP-activated protein kinase (AMPK), protein kinase B (AKT), or mitogen-activated protein kinase (MAPK) activities, which phosphorylate mTOR and inactivate the mTOR1 complex (mTORC1). Inactive mTOR dissociates and activates the unc-51-like kinase 1 (ULK1) complex that, in turn, binds and activates the class III phosphatidylinositol 3–kinase complex I (PI3KC3-C1), which is recruited on the phagophore assembly site (initiation stage, see[32] for a comprehensive review of autophagy initiation). Here, the engaged complexes orchestrate the fusion of plasma membranes derived from endoplasmic reticulum, Golgi apparatus, or endosomes to finally generate the phagophore. In particular, the PI3KC3-C1 complex, that includes Beclin-1 (BECN1), autophagy-related 14 (ATG14), vacuolar protein sorting 15 (VPS15), and VPS34, locally produces phosphatidylinositol 3-phosphate (PI3P) (nucleation stage)[33] where lipid-binding proteins double FYVE domain-containing protein 1 (DFCP1) and WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) are recruited and create a platform for assembly and activation of several autophagy-related proteins such as ATG12, ATG7, ATG10, ATG5, and ATG16L1, with an ubiquitin-like activity[34][35]. This sequential recruitment of autophagy factors on lipid domains initially produces the omegasome, membrane platforms connected to the endoplasmic reticulum[34], and culminates with the conjugation of another ubiquitin-like protein, microtubule-associated protein 1A/1B-light chain 3 (LC3-I), with the lipid phosphoatidylethanolamine (PE). Lipidated LC3 (LC3-II) is then anchored on the nascent phagophore where it mediates elongation of the phagophore membrane[36] (elongation stage). LC3-II is widely recognized as a key marker of autophagosome formation and autophagic activity, since lipidated LC3 targets autophagosome membranes and is then degraded after fusion with lysosome[37]. Finally, autophagosome closure is mediated by a component of the endosomal sorting complexes required for transport (ESCRT), CHMP2A (charged multivesicular body protein 2A), which regulates the formation of the double-membrane autophagosome through AAA-ATPase VPS4 activity[38]. The formed autophagosome sequesters cytosolic components and fuses with lysosomes (instigating the autolysosome) where autophagosome cargoes are exposed to the hydrolytic actions of lysosomal hydrolases (see[39] for a detailed review of autophagosome–lysosome fusion) (Figure 1). In selective autophagy, several adaptor proteins link payloads to the autophagosome, including the autophagic receptor p62 (or sequestosome 1, SQSTM1,) that binds cargoes (mostly ubiquitinated proteins) to LC3 in elongating phagophore[40]. Since p62 is itself degraded by autophagy, its expression levels are another canonical marker to monitor the autophagic flux[37]. For an efficient fusion, the autophagosome migrates to lysosome locations, predominantly found in perinuclear region, aided by the molecular adaptor Ras-related protein Rab-7a (Rab7a) that links the autophagosome to a microtubule motor[41]. The minus-end-directed dynein−dynactin motor complex transports the autophagosome close to a lysosome[42] where Rab proteins and tethering complexes drive cognate vesicle-soluble NSF attachment protein receptors (SNAREs) in autophagosome and lysosome to mediate the fusion of the two cellular compartments[43].
3. The Ideal Outcome of Inflammation: Resolution
3.1. Key Aspects of Acute Inflammation and Resolution
Acute inflammation is a tightly regulated and self-limited protective reaction against dangerous materials, expected to eliminate the insulting stimulus and to restore tissue function[44]. After injuries on vascularized tissues, the increase in endothelial permeability results in plasma leakage (edema) and a rapid PMN recruitment to remove the inflammatory stimulus by phagocytosis, degranulation, or formation of neutrophil extracellular traps (NETs)[45]. PMNs then undergo apoptosis and are removed through efferocytosis by MФs differentiated from blood monocytes[46]. In addition to efferocytosis, MФs are also able to remove the inflammatory trigger by non-phlogistic phagocytosis[47]. Successful efferocytosis then stimulates MФ skewing from pro-inflammatory M1 to pro-resolving M2 phenotypes that dampen inflammation while stimulating tissue repair[48]. Once tissue homeostasis is restored, MФs exit the inflamed tissue by the lymphatic system.
These cellular events are timely and spatially regulated by the concerted production of proteins and lipids released by effector and resident cells[49]. Early in inflammation, pro-inflammatory eicosanoids (mainly prostaglandins-PGs and leukotrienes-LTs) derived from arachidonic acid (AA) metabolism), inflammatory cytokines and chemokines are produced by tissue-resident cells after sensing injuries like pathogen infection. These mediators in turn regulate blood flow, induce vasodilatation, edema formation and PMN infiltration, to mount the inflammatory reaction. Despite PMN recruitment is crucial to restrict the noxious stimulus, an excessive and uncontrolled PMN infiltration, activation and release of antimicrobial products may be harmful to the tissue as well as the prolonged secretion of pro-inflammatory mediators. Therefore, a resolution program should be started[50]. A crucial step in resolution is a lipid mediator class switch, from pro-inflammatory eicosanoids to SPMs that include lipoxin (LX), resolvins (Rv), maresins (Mar), and protectins (PD) (for a recent review on SPMs readers are referred to[51]). These are biosynthesized from essential polyunsaturated fatty acids (PUFAs) (see below) and act by limiting PMN infiltration, regulating the balance between pro-inflammatory and anti-inflammatory cytokines and chemokines, and by stimulating phagocytosis, efferocytosis, killing and wound healing by MФs[11]. Notably, SPM biosynthesis can be modulated by pro-inflammatory mediators, for instance PGE2, generated during the onset of the inflammatory response[52], indicating that resolution is programmed from the beginning of the inflammatory reaction. Figure 2 illustrates an integrated view of inflammation and its resolution.
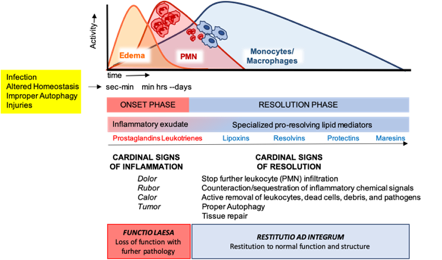
Figure 2. The acute inflammatory response and its ideal outcome: essential steps, mechanisms, and definitions. Injury, infections, or dysregulated homeostasis ignite the acute inflammatory response that is normally a host protective mechanism. The first event in acute inflammation is edema formation, followed by infiltration of polymorphonuclear leukocytes (PMNs), and then monocyte and macrophages that clear PMNs leading to resolution, which is essential for ensuring host protection and sparing from tissue damage.
3.2. SPM Biosynthesis
Using a system approach of spontaneous and self-contained inflammation, pioneering studies from Dr Serhan’s laboratory lead to the identification of several previously undescribed lipid compounds in inflammatory exudates during the resolution phase[53]. These mediators act locally on different cell types by exploiting several receptors in a spatial- and temporal-dependent manner.
LXA4 and B4, the first SPMs identified[54], mainly derive from transcellular metabolism of AA involving PMN 5-lipoxygenase (LO) and platelet 12-LO[55][56]. In a second pathway of LX biosynthesis, AA is metabolized by the sequential activity of 15-LO in epithelial cells or MФs and leukocyte 5-LO[57][58]. A third route of LX generation involves aspirin-acetylated cyclooxygenase (COX)-2 in vascular and epithelial cells and leukocyte 5-LO. LX produced by this metabolic pathway are isomers of native LX termed aspirin-triggered LX (ATL) A4 and B4 [59].
D-series Rv (RvD) encompass six metabolites (RvD1-6) of the omega-3 fatty acid docosahexaenoic acid (DHA) differing from number, position, and chirality of their hydroxyl residues, and position and isomerism of their double bonds[60]. The four members of E-series Rv (RvE1, RvE2, RvE3, and 18S-Rv1) are metabolites of the omega-3 fatty acid eicosapentaenoic acid (EPA) generated, similarly to ATL, through aspirin-acetylated endothelial COX-2 and PMN 5-LO transcellular activity[61]. More recently, the structure and biosynthesis of four members of the T-series Rv – which carry a 13-C position -OH group - have been elucidated. Specifically, similarly to RvE, RvT1-4 are produced from transcellular metabolism involving aspirin-acetylated endothelial COX-2 and PMN 5-LO but using n-3 docosapentaenoic acid (DPA) as precursor[62].
PDs are metabolites of DHA produced by PMNs, MФs, and eosinophils, following enzymatic activity of 15-LO (PD1 or neuroprotectin when produced in neural system) or aspirin-acetylated COX-2 (AT-PD1) and subsequent enzymatic epoxidation and hydrolysis. PD1 isomerization leads to the production of a less-characterized PD member, PDX[63].
Mar1 and Mar2 are a third family of SPMs produced from DHA at sites of inflammation via a 12-LO enzymatic activity in MФs[64], probably produced later during inflammation following the appearance of resolution phase MФs.
In addition, recent work has uncovered additional SPMs showing potent tissue-protective activities in addition to their pro-resolution roles, and termed SPM conjugated in tissue regeneration (CTR). Characterization of their structural properties showed that CTRs originate from DHA and contain sulfido-conjugates derived from glutathione conjugation to DHA[65]. Maresins CTR (MCTR1-3)[66], resolvins CTR (RTCTR1-3) and protectins CTR (PCTR1-3)[65] are members of this class of SPMs. Finally, in addition to RvT, n-3 DPA is also the substrate of other SPMs with potent anti-inflammatory activities named PD1n-3, RvD1n-3, RvD2n-3, RvD5n-3, MaR1n-3, MaR2n-3, and MaR3n-3 based on their structural analogies to PD, RvDs, and Mar, respectively[67].
3.3. SPM Receptors and Functions in Resolution of Inflammation
SPMs regulate inflammation and resolution via activation of different cell surface G-protein-coupled receptor (GPCRs) (reviewed in[68]), which rapidly transport signals and activate intracellular pathways to control a number of biological functions. Several SPM receptors have been identified to date mostly using library screening, labelled ligands for specific binding, GPCR–β-arrestin-coupled system and functional cellular responses involving gain and loss of function approaches[69][70][71].
Despite Formyl peptide receptor 2 (ALX/FPR2, also termed FPR2, FPRL1, or FPR2/ALX) being the first discovered and the most studied SPM receptor able to convey LXA4 bioactions in PMNs[72], it is now clear that this receptor is expressed in multiple cell types including myeloid cells and lymphocytes, resident endothelial, epithelial cells, fibroblasts[73], and stem cells[74], and is also activated by other SPMs (RvD1, AT-RvD1, RvD3, AT-RvD3, and AT-LXA4)[69][75][76][77][78]. In addition, in vitro studies showed that ALX/FPR2 is a promiscuous receptor that is also recognized and stimulated by a variety of natural and synthetic peptides to translate effects ranging from pro-inflammatory to anti-inflammatory and pro-resolution depending on the activating ligand (reviewed in[79]). However, the ALX/FPR2 transgenic and knock-out mouse models confirmed the crucial role of this receptor in inflammation resolution[80][81], and findings in human indicated that ALX/FPR2 levels dictate the amplitude of inflammation and its defective expression could be associated with inflammatory disorders[82][83].
In addition to ALX/FPR2, RvD1, AT-RvD1, RvD3, AT-RvD3, and AT-LXA4 also activate the human DRV1/GPR32 receptor[69][77]. Furthermore, RvD5 was also reported as agonist for DRV1/GPR32[76].
RvD2 exerts its pro-resolving activities by binding and activation of the DRV2/GPR18 receptor in PMNs, monocytes and MФs[84], thus stimulating bacterial clearance and organ protection in infectious inflammation[85].
ERV/ChemR23 is a GPCR very similar to ALX/FPR2 that binds and mediates RvE1 signals in monocytes, dendritic cells, and MФs[70][71]. In addition, RvE1 also interacts with BLT1, the LTB4 receptor, as partial agonist, to dampen LTB4 pro-inflammatory signals while stimulating resolution through ERV/ChemR23[86].
Similarly, Mar1 partially interacts with recombinant human BLT1 and acts as full agonist for the leucine-rich repeat containing G protein–coupled receptor 6 (LGR6) in MФs to increase phagocytosis and efferocytosis of human and mouse phagocytes[87].
More recently, GPR37 was identified as potential receptor involved in PD1-mediated MФs phagocytosis and resolution of inflammatory pain[88] and GPR101 proved to mediate pro-resolving actions of RvD5n-3 DPA[89].
On the other hand, if and how T-series Rv and the other 3-DPA metabolites and CTRs activate similar or different GPCRs is currently unknown.
At the molecular level, despite SPM/receptor interaction is cell- and organ-specific and influences a variety of pathways related to inflammation, some common features emerge. For example, most SPMs induce receptor dimerization, dampen nuclear factor kappa-light-chain-enhancer of activated B cells( NF-κB)[90] and Nrf-2[18] activation, modulate the ERK, EGFR, and mTOR pathways[91], and regulate the expression of inflammatory microRNAs (miRNAs)[92]. At the cellular level, SPMs limit PMN recruitment and activity; enhance MФ phagocytosis and efferocytosis; counter-regulate production of pro-inflammatory mediators, including PGs and LTs; chemokines and cytokines such as tumor necrosis factor a (TNFα), interleukin (IL)-6 and IL-8; growth factors (vascular endothelial growth factor, VEGF) and reactive oxygen species, whereas stimulating the production of nitric oxide (NO) and PGI1 by endothelial cells (recently reviewed in[93]). Since non-resolving inflammation underlies the pathogenesis of many human diseases and given their potent bioactions in cellular and preclinical models[12], SPMs are now being tested in selected human diseases with promising outcomes[23][24][25].
4. Autophagy in Chronic Inflammatory Diseases
4.1. Autophagy and Inflammation
Increasing evidence indicates that autophagy plays crucial roles in the regulation of the inflammatory response. Therefore, dysregulation of autophagy may contribute to trigger and amplify chronic inflammation and could be an underlying mechanism of human chronic inflammatory disorders.
As discussed above, the main function of autophagy is to maintain cellular functions under stress conditions, in order to prevent cell death. In addition to cell homeostasis, autophagy is centrally involved in host defense. Indeed, autophagy could be activated by invading microorganisms that trigger metabolic stress and inhibit mTOR, or by direct recognition of pathogens through innate immune receptors (toll like receptors—TLRs, NOD-like receptors—NLRs, and RIG-like receptors) or autophagic adaptors (sequestosome 1-like receptors—SLRs)[94]. Autophagy contributes to pathogens elimination by two mechanisms: sequestration and degradation of intracellular microorganisms by xenophagy[95]; decoration with LC3 molecules of pathogens captured by phagocytosis to promote phagosomal maturation and phagolysosomes formation (LC3-associated phagocytosis—LAP)[96].
Autophagy controls inflammation also by regulating the production of inflammatory mediators. The NLR family pyrin domain containing 3(NLRP3) inflammasome is a multiprotein complex able to sense exogenous or endogenous damage and to produce the pro-inflammatory IL-1β and IL-18, thus inducing inflammation and pyroptosis[97]. Recent evidence shows that damaged mithocondria stimulate NRLP3 inflammasome activation[98] and that damaged mithocondia are removed by mithopagy[99]. Therefore, autophagy dysfunction impairs mithocondria removal, resulting in leakage of endogenous NLRP3 agonists, i.e. mithocondrial DNA and reactive oxygen species (ROS), and excessive production of IL-1β and IL-18 that sustain unrelenting inflammation[100]. Importantly, autophagy may down-regulate excessive inflammasome activity in the effort to dampen exaggerate inflammation[101], again suggesting that failure in autophagy may fail to counter-balance inflammation. In addition, autophagy negatively regulates type I interferon (IFN-I) production in response to DNA or RNA viruses (recently reviewed in[102]). For example, following detection of cytosolic viral DNA, BECN1, ATG9A, and ULK1 halt downstream signaling, leading to IFN-I production and anti-viral responses[103][104][105]. Similarly, viral RNA in cytosol stimulates RIG-I-dependent pathways that induce IFN-I production by mithocondria. This pathway is inhibited by the recruitment of ATG12-ATG5-ATG16L1 autophagic complex on mithocondrial membrane with suppression of ROS-mediated IFN-I release[106][107].
In addition to finely-tune the production of selected cytokines, autophagy may also regulate NF-κB activity, a key transcription factor of the inflammatory response[108]. Early work shows that p47 negatively regulates NF-κB activity by targeting NF-kappaB essential modulator (NEMO), a component of the IκB kinase (IKK) complex that stimulates NF-κB translocation to the nucleus[109], for lysosomal degradation[110]. Therefore, p47 emerges as an autophagic receptor to selectively promote NEMO degradation by autophagy. Furthermore, the F-box protein SKP2 acts as a bridge to promote the interaction between IKKb, a crucial kinase upstream NF-κB activation, and the autophagic receptor p62. This interaction finally leads to IKKb degradation via selective autophagy and reduction of NK-kB activity[111]. Similarly, the angiopoietin-like 8 (ANGPTL8) protein bridges IKKg with p62 and promotes IKKg degradation by selective autophagy, thereby negatively regulating NF-κB activation and inflammation[112]. Therefore, failure in selective autophagy may promote inflammation by circumventing regulatory loop that controls NF-κB activity.
Autophagy also crucially regulates important activities of PMNs and MФs[113]. Indeed, autophagy is activated in PMNs and is necessary for PMN functions such as differentiation[114], bacterial clearance, degranulation and ROS production. Indeed, pharmacological and genetic approaches demonstrated that autophagy impairment reduces bacterial killing by human PMNs[115]. Along these lines, autophagy inhibition also reduces NETs formation[116], ROS production by inhibition of NADPH oxidase, and degranulation through altered fusion of PMN granules and phagosomes[117]. Collectively, these results suggest that autophagy inhibition may reduce the PMN-driven inflammatory reaction. Similarly, several monocytes/MФs functions are crucially regulated by autophagy[118]. For instance, during monocyte-to-MФ differentiation, ULK1 activation and BECN1 dissociation from inhibitory B-cell lymphoma 2 (BCL2) trigger autophagy and promote differentiation and generation of functional phagocytes[119][120]. In addition, autophagy regulates almost every aspect of MФ biology, from pathogen recognition to cytokine release, inflammasome activation, and polarization. For further details please refer to a recent review on this area[121].
Moreover, autophagy plays critical functions in adaptive immunity. This topic has been discussed in some excellent reviews[7][122][123] and will not be further discussed here since it would be beyond the scope of this article.
Since autophagy is a key player of the inflammatory response, is not surprising that a number of chronic inflammatory human diseases are characterized by deregulated autophagy. Key aspects of some paradigmatic examples are listed below.
4.2. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Crohn’s Disease
Crohn’s disease (CD) was the first human pathology where a significant pathogenetic contribution of autophagy has been elucidated. CD is a chronic inflammatory bowel disease characterized by excessive inflammation and impaired bacterial clearance[124]. Genome-wide association studies revealed single nucleotide polymorphisms in autophagy genes, in particular ATG16L1, which is involved in autophagosome formation; immunity-related GTPase family M protein (IRGM) that regulates autophagy through mitochondria interaction; and the bacterial sensor, NOD2[125][126]. Functional studies then revealed that defects in autophagy lead to impaired clearance of bacteria, altered production of pro-inflammatory cytokines[127], and defects in granule production by Paneth cells[128]. Importantly, Atg16l1 deficiency in mouse PMNs and MФs increased ROS production but impaired microbial killing and altered IL-1β release of by MФs[127][129], further documenting that defects in autophagy exacerbate bowel inflammation.
4.3. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Cystic Fibrosis
Autophagy dysfunction is critically involved in several pulmonary diseases characterized by non-resolving inflammation[130]. Cystic fibrosis (CF) is a genetic disorder caused by mutations (deletion of phenylalanine from the position-508—F508del is the most common) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene[131], a membrane ion channel primarily regulating chloride efflux. Non-resolving lung inflammation in CF impairs host defense and is characterized by an uncontrolled recruitment of PMNs that, once infiltrated, release toxic products such as elastase, oxidant species, and NETs that cause tissue damage and mucus plugging. Evidence indicates that CFTR mutations stimulate a pro-inflammatory phenotype, even independently from infection[132]. Indeed, CF airways contain elevated concentrations of soluble mediators, including IL-8, -6, and LTB4 that contribute to the progression of lung disease[133]. Moreover, the reduced formation of SPMs may also contribute to CF relentless inflammation, suggesting that SPM administration could provide clinical benefits[19][20][21][134][135].
In addition to SPM defects, recent results point to autophagy as to a potential contributor to the pathogenesis of exuberant inflammation in CF. Indeed, human airway epithelial cells show accumulation of polyubiquinated proteins, impaired autophagy, and failure in the clearance of aggresome by aggrephagy. At the molecular level, defective CFTR causes the displacement of BECN1 from the endoplasmic reticulum and drives the sequestration of the PI3KC3 complex in aggresomes[120]. As a consequence, autophagy is disabled leading to defective autophagosome formation, accumulation of p62, and increased aggresome, which further sequester defective F508del CFTR[120]. Importantly, reconstitution of BECN1 restores CFTR trafficking to plasma membrane, reduces aggresome formation, and dampens production of pro-inflammatory mediators by CF cells, thus establishing a clear link between defective autophagy and sustained inflammation[136]. In addition, autophagic defects seem to be also related to the dysregulated NLRP3 inflammasome activity in CF. Indeed, administration of anakinra, an IL-1 receptor antagonist, to mouse and human CF models suppresses NLRP3-mediated inflammation and activates autophagy, suggesting that correction of autophagy could downregulate excessive inflammasome activity and thereby restore an appropriate inflammatory response[137]. Along these lines, treatment with the anti-inflammatory peptide IDR-1018 also attenuated hyperinflammatory cytokine secretion, restoring an appropriate autophagic flux in CF cells[138].
Importantly, rescuing autophagy also has beneficial effects against intestinal inflammation and dysfunction in patients with CF[139], thus extending the importance of autophagy as regulator of inflammation in multiple districts in CF.
In addition to epithelia, autophagy dysfunction also significantly contributes to defects of innate immunity in CF. Indeed, CF MФs show weak autophagic activity, partially due to hypermetilation of the Atg12 promoter[140] and to elevated levels of the autophagy-regulating miRNAs miR-17 and miR-20a[141]. Stimulation of autophagy by the mTOR inhibitor rapamycin[142] or depletion of overexpressed p62[143] reestablishes the ability of CF MФs to clear Burkholderia cenocepacia infections and ameliorates lung inflammation. Similarly, autophagy inhibition improves the ability of mast cells to clear Pseudomonas aeruginosa, while pharmacologic induction of autophagy increases bacterial clearance in lungs of CF mice[144]. Along the same lines, treatment with the antibiotic azithromicin prevents lysosomal acidification and autophagosomal degradation, decreasing intracellular killing of multi-drug resistant Mycobacteria abscessus by MФs, thus triggering chronic infection and inflammation[145]. Therefore, besides epithelia, autophagic deficiency also impairs the ability of immune cells to clear infections, thus sustaining chronic inflammation.
Collectively, these results indicate that CFTR deficiency causes an inhibition of autophagy in multiple cells and distinct organs in CF, suggesting that pharmacological induction of autophagy may provide benefits. Indeed, in vivo studies with CF mice, human CF primary cells, and patients with CF show that cysteamine plus epigallocatechin gallate (EGCG) restore BECN1-dependent autophagy, rescue functional F508del CFTR on the plasma membrane of nasal epithelial cells, and reduce pro-inflammatory cytokines in sputum of patients with CF[146][147]. These effects may be improved using the recently introduced combination cysteamine-amiodarone as autophagy inducers[148]. Other potential pharmacological treatments for CF based on autophagy correction were recently reviewed by Bodas et al.[149]. Although none of these treatments have yet reached the bedside, probably due to the high doses required and for the many off-target effects, approaches to restore autophagy and resolution of inflammation in CF can be potentially relevant.
4.4. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Cancer
Experimental and clinical evidence indicates that chronic, non-resolving inflammation contributes to the development and diffusion of neoplasms in humans[150][151]. Indeed, early events in tumor progression are characterized by altered production of inflammatory mediators and recruitment of inflammatory cells, i.e., PMNs and MФ, to support tumorigenesis[151].
Likewise, altered autophagy is also a key driver of malignant transformation with a peculiar dual role depending on cancer stage. Indeed, in early steps of cancer development, autophagy works as a tumor suppressor pathway through removal of damaged organelles, proteins, ROS, and p62, which otherwise would sustain tumorigenesis and genomic instability. On the contrary, in established tumors, autophagy activation fuels the increased metabolic requirement of cancer cells by providing nutrients and energy that sustain tumor growth (recently reviewed in[152]). In addition, autophagy impacts on tumor development also by exploiting the inflammatory response to support the implant, growth, and pharmacological response of tumors.
For example, oncogenic viruses have developed clever mechanisms to revert control of cell proliferation and to trigger oncogenesis through manipulation of autophagy[153][154]. In fact, human papillomaviruses (HPV) down-regulate the autophagic response of infected keratinocytes in cervical and head and neck cancers to extend host’s cell lifespan[153]. In addition, recent work indicates that the HPV16 E7 oncoprotein manipulates autophagy to dampen the host immune reaction against infected cells. Indeed, E7 exploits autophagy to promote the degradation of STING, an immune signaling complex that stimulates anti-viral type I IFN production, thus impairing host immunity against HPV infections[155][156].
In addition, autophagic defects in mouse models of pancreatic cancer trigger the expression of pro-inflammatory cytokines such asChemokine (C-C motif) ligand 5 (CCL5) and IL-6, and increase the infiltration of PMNs and T-cells, which fuel dysplastic transformation and tumor initiation[157]. Therefore, this observation indicates that autophagy restrains tumor induction by dampening inflammation. Along these lines, a proteomic investigation of Ras-driven cancer cells highlights that autophagy selectively targets specific inflammatory mediators such as IL-6, IFN-α, and IFN-β to suppress inflammation, and that autophagic defects activate innate immunity and the interferon response[158]. Consistent with this, autophagic-deficient tumor-bearing mice show increased expression of genes involved in the inflammatory response, leukocyte migration and immune cell trafficking as well as enhanced intertumoral MФ infiltration, which sustain cancer progression and increase mortality[144][145]. Together, these results support the notion that defective autophagy in cancer promotes chronic inflammation that, in turn, sustains cancer development.
Importantly, autophagy and inflammation play a crucial role also during response to therapy. In fact, depletion of essential autophagic genes such as Atg5 or Atg7 in mouse allografts blunts the recruitment of immune cells, indicating that immunogenic response against cancer is crucially regulated by the autophagy-inflammation crosstalk[159][160]. Additionally, ATG7 siRNA in thyroid cancer cell lines sensitizes resistant cell lines to immune cell-mediated cytotoxicity[161]. Therefore, downregulation of autophagy during treatments may potentiate the immune reaction against residual tumor cells in selected cancers.
4.5. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Alzheimer’s Disease
Alzheimer’s Disease (AD) is one of the most common neurodegenerative disorder that is characterized by cognitive decline and the presence of amyloid β plaques and neurofibrillary tangles[162]. Over the last decade, a sustained immune response in the brain has emerged as a key pathogenetic feature in AD. Indeed, the chronic presence of highly insoluble deposits of amyloid β and tau and other inflammatory mediators may determine a functional impairment of microglia, the brain-resident MФs-type cell, leading to chronic neuroinflammation and consequent neuronal degeneration and synaptic dysfunction[163]. Emerging evidence indicates that interplays between inflammation and autophagy may occur in the AD brain. Chronic inflammation determines intracellular ROS accumulation that, with aging, becomes progressively detrimental by triggering inflammasome formation[164], as well as apoptotic pathways and mitochondrial dysfunction[165]. These pathways induce autophagic mechanisms to suppress local damage[166]. More specifically, mitochondria impairment stimulates mitophagy that is impaired in the hippocampus of AD patients. Mitophagy stimulation reverses memory impairment through PINK-1 (PTEN-induced kinase-1), PDR-1 (Parkinson’s disease- related-1; parkin), or DCT-1 (DAF-16/FOXO-controlled germline-tumor affecting-1)-dependent pathways. Moreover, mitophagy diminishes insoluble Aβ1–42 and Aβ1–40 and prevents cognitive impairment in an APP/PS1 mouse model through microglial phagocytosis of extracellular Aβ plaques and suppression of neuroinflammation[167].
5. Stimulation of Resolution Restores Autophagy and Dampens Chronic Inflammation
It is now clear that the pro-resolving and the autophagic circuits can interact at several junctures, making the analysis of such interplay very appealing in the perspective of developing new and more potent anti-inflammatory pharmacology to combat a variety of chronic, life-threatening human diseases.
The first evidence that SPMs can activate autophagy comes from an elegant study showing autophagy induction by RvD1 and AT-LXA4 in human and murine monocytes and MФs[168]. Treatment with nanomolar concentrations of these SPMs triggers LC3-I to LC3-II formation, increases autophagosome formation while decreasing p62 levels, suggestive of autophagy activation. Notably, RvD1 and AT-LXA4 activate autophagy through a MAPK1 pathway, independently from mTOR, by inducing the dissociation of BCL2 from BECN1, with subsequent BECN1 activation and autolysosome formation. Importantly, RvD1 and AT-LXA4 stimulate phagocytosis of zymosan particles by MФs in an ATG5-dependent manner[168], suggesting that autophagy activation is crucial for the removal of inflammatory stimuli, to promote resolution and avoid chronic inflammation.
Along these lines, an agonist of the ALX/FPR2 receptor, BML-111, promotes LC3-II accumulation in alveolar MФs primed with LPS to mimic acute lung injury, suggestive of increased autophagosome formation. In addition, reduction of p62 levels confirms that stimulation of ALX/FPR2 by BML-111 activates autophagic flux through a MAPK pathway independent from mTOR, suggesting late autophagic activation. Interestingly, BML-111 administration to rats with acute lung injury reduces the concentration of pro-inflammatory cytokines in lung lavage and activates autophagy in alveolar MФs, supporting the hypothesis that enhanced autophagy dampens inflammation[169].
Another ALX/FPR2 agonist, RvD1, proved beneficial in restoring autophagic flux in a mouse model of cerulein-induced acute pancreatitis (AP), where autophagy plays a clear role in supporting inflammation[170]. Indeed, AP tissues are characterized by impaired autophagy with higher expression of BECN1, p62, LC3-II, and increased number of autophagic vacuoles. Treatment with RvD1 normalizes the autophagic flux in AP mice by reducing BECN1, p62, LC3-II levels, and the number of autophagic structures[171].
Similarly to RvD1, RvE1 has been recently reported as modulator of doxorubicin-induced autophagy in cardiac tissues. Indeed, administration of RvE1 to doxorubicin-treated mice restores normal levels of BECN1, p62, and LC3-II in heart by an AKT/mTOR-dependent signaling pathway, suggesting that RvE1 protects from cardiotoxicity by modulating autophagy[172].
Opposite to RvD1 and RvE1, LXA4 inhibits obesity-induced autophagy in mice fed with a high-fat diet[173]. Chronic obesity promotes excessive autophagy which, in turn, sustains cell death and adipose inflammation[174]. Treatment with LXA4 or its stable analog benzo-LXA4increases p62 and reduces LC3-II expression in adipose tissue of high-fat diet animals as compared to standard-diet littermates, indicative of autophagic flux suppression. Importantly, this activation is independent from both mTOR and AMPK activity[173], suggesting that LXA4 may regulate a late stage of autophagosome formation, consistently with previous results[168]. Similarly to LXA4, Mar1 has been reported as an autophagy inhibitor in models of obesity. Indeed, treatment of 3T3-L1 adipocytes with a pro-inflammatory stimulus such as TNF-α reduces p62 and increases LC3-II levels, suggestive of autophagy activation, and these effects are reverted in a concentration-dependent manner by pre-treatment with Mar1[175]. However, Mar1 effects on autophagy may vary with the biological setting. Indeed, in models of chronic periodontitis, Mar1 activates autophagy in human periodontal ligament cells (PDL) exposed to LPS, as suggested by the upregulation of LC3-II and BECN1, and the downregulation of p62 in Mar1-treated PDL[161]. Notably, the autophagic inhibitor 3-MA antagonizes the inhibitory actions of Mar1 on IL-6, IL-8, TNF-α, and IL-1β release by PDL exposed to LPS[176], suggesting that Mar1 limits inflammation by activating autophagy. Moreover, Mar1 has been reported to stimulate autophagy in a mouse model of AD, where in addition to improve cognitive decline and to balance the production of pro-inflammatory and anti-inflammatory mediators, it increases BECN1 and LC3-II levels and decreases p62 in the hippocampus, indicative of autophagy stimulation[177].
In addition to SPMs, some of their precursors proved effective in autophagy modulation. In particular, in hepatocytes primed with palmitate to mimic low grade chronic inflammation, lipotoxicity, and insulin resistance associated with metabolic disorders in obesity[178], the DHA-metabolite 17(S)-HDHA restores autophagy as demonstrated by LC3-II and p62 accumulation in cells with blocked autophagic flux[179].
The following Table 1 and Figure 3 summarize the role of SPMs as modulators of autophagy and inflammation in several models of human diseases.
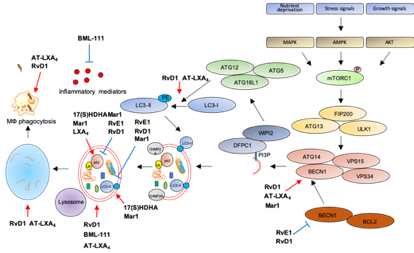
Figure 3. Modulation of autophagy by SPMs. Red arrows: increase; blue bar-headed arrows: decrease in indicated proteins or subcellular compartments.
Table 1. Role of SPMs as modulators of autophagy and inflammation in human diseases.
|
SPMs |
Disease Model |
Molecular Effect |
Effect on Autophagy |
Refs |
|
RvD1 and AT-LXA4 |
MФs inflammation |
BECN1 dissociation and activation MAPK dependent, autolysosome formation |
Activation |
[168] |
|
BML-111 |
Acute lung injury, impaired autophagy |
LC3-II accumulation, p62 degradation MAPK dependent |
Activation |
[169] |
|
RvD1 |
Acute pancreatitis, impaired autophagy |
Reduction in BECN1, p62, LC3-II |
Reactivation of impaired autophagic flux |
[171] |
|
RvE1 |
Cardiotoxicity, impaired autophagy |
Reduction in BECN1, p62, LC3-II |
Reactivation of impaired autophagic flux |
[172] |
|
LXA4 |
Obesity, increased autophagy |
LC3-II decrease, p62 increase |
Inhibition |
[173] |
|
Mar1 |
Obesity, increased autophagy |
LC3-II decrease, p62 increase |
Inhibition |
[175] |
|
17(S)-HDHA |
Obesity, increased autophagy |
LC3-II and p62 accumulation in autophagic flux experiments |
Inhibition |
[179] |
|
Mar1 |
Periodontitis |
Increase in LC3-II and BECN1, decrease in p62 |
Activation |
[176] |
|
Mar1 |
Alzheimer, impaired autophagy |
Increase in LC3-II and BECN1, decrease in p62 |
Activation |
[177] |
References
- Christian De Duve; Robert Wattiaux; Functions of Lysosomes. Annual Review of Physiology 1966, 28, 435-492, 10.1146/annurev.ph.28.030166.002251.
- Takahiro Shintani; Daniel J. Klionsky; Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990-995, 10.1126/science.1099993.
- Arnold, J.; Murera, D.; Gros, F. Autophagy in Chronic Inflammation. In Autophagy Networks in Inflammation; Maiuri, M.C., De Stefano, D., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 107–133. ISBN 978-3-319-30077-1.
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101.
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260.
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24.
- Vojo Deretic; Tatsuya Saitoh; Shizuo Akira; Autophagy in infection, inflammation and immunity. Nature Reviews Immunology 2013, 13, 722-737, 10.1038/nri3532.
- Mihai G. Netea; Frances Balkwill; Michel Chonchol; Fabio Cominelli; Marc Y. Donath; Evangelos J. Giamarellos-Bourboulis; Uglas Golenbock; Mark S. Gresnigt; Michael T. Heneka; Hal M. Hoffman; et al.Richard HotchkissLeo A.B. JoostenDaniel L. KastnerMartin KorteEicke LatzPeter LibbyThomas Mandrup-PoulsenAlberto MantovaniKingston H. G. MillsKristen L. NowakLuke A. O’NeillPeter PickkersTom Van Der PollPaul M. RidkerJoost SchalkwijkDavid A. SchwartzBritta SiegmundClifford J. SteerHerbert TilgJos W.M. Van Der MeerFrank L. Van De VeerdonkCharles A. Dinarello A guiding map for inflammation. Nature Immunology 2017, 18, 826-831, 10.1038/ni.3790.
- David Furman; Judith Campisi; Eric Verdin; Pedro Carrera-Bastos; Sasha Targ; Claudio Franceschi; Luigi Ferrucci; Derek W. Gilroy; Alessio Fasano; Gary W. Miller; et al.Andrew H. MillerAlberto MantovaniCornelia M. WeyandNir BarzilaiJorge GoronzyThomas A. RandoRita B. EffrosAlejandro LuciaNicole C. KleinstreuerGeorge M. Slavich Chronic inflammation in the etiology of disease across the life span. Nature Medicine 2019, 25, 1822-1832, 10.1038/s41591-019-0675-0.
- Charles N. Serhan; Nan Chiang; Thomas E. Van Dyke; Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology 2008, 8, 349-361, 10.1038/nri2294.
- Charles N. Serhan; Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92-101, 10.1038/nature13479.
- Georg Andreas Schett; Markus F. Neurath; Resolution of chronic inflammatory disease: universal and tissue-specific concepts.. Nature Communications 2018, 9, 3261, 10.1038/s41467-018-05800-6.
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63.
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67.
- Lee, C.-T.; Teles, R.; Kantarci, A.; Chen, T.; McCafferty, J.; Starr, J.R.; Brito, L.C.N.; Paster, B.J.; Van Dyke, T.E. Resolvin E1 Reverses Experimental Periodontitis and Dysbiosis. J. Immunol. 2016, 197, 2796–2806.
- Li, C.; Wu, X.; Liu, S.; Shen, D.; Zhu, J.; Liu, K. Role of Resolvins in the Inflammatory Resolution of Neurological Diseases. Front. Pharmacol. 2020, 11, 612.
- Lucy Victoria Norling; Sarah E. Headland; Jesmond Dalli; Hildur H. Arnardottir; Oliver Haworth; Hefin R. Jones; Daniel Irimia; Charles N. Serhan; Mauro Perretti; Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight 2016, 1, null, 10.1172/jci.insight.85922.
- Alessandro Matte; Antonio Recchiuti; Enrica Federti; Bérengère Koehl; Thomas Mintz; Wassim El Nemer; Pierre-Louis Tharaux; Valentine Brousse; Immacolata Andolfo; Alessia Lamolinara; et al.Olga WeinbergAngela SicilianoPaul C. NorrisIan R. RileyAchille IolasconCharles N. SerhanCarlo BrugnaraLucia De Franceschi Resolution of sickle cell disease–associated inflammation and tissue damage with 17R-resolvin D1. Blood 2019, 133, 252-265, 10.1182/blood-2018-07-865378.
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 2018, 11, 35–49.
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252.
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581
- Gabrielle Fredman; Ira Tabas; Boosting Inflammation Resolution in Atherosclerosis. The American Journal of Pathology 2017, 187, 1211-1221, 10.1016/j.ajpath.2017.01.018.
- Kong, X.; Wu, S.-H.; Zhang, L.; Chen, X.-Q. Pilot application of lipoxin A4 analog and lipoxin A4 receptor agonist in asthmatic children with acute episodes. Exp. Ther. Med. 2017, 14, 2284–2290.
- Wu, S.H.; Chen, X.Q.; Liu, B.; Wu, H.J.; Dong, L. Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br. J. Dermatol. 2013, 168, 172–178.
- Motwani, M.P.; Colas, R.A.; George, M.J.; Flint, J.D.; Dalli, J.; Richard-Loendt, A.; De Maeyer, R.P.H.; Serhan, C.N.; Gilroy, D.W. Pro-resolving mediators promote resolution in a human skin model of UV-killed Escherichia coli–driven acute inflammation. JCI Insight 2018, 3, e94463.
- Masahide Oku; Yasuyoshi Sakai; Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays 2018, 40, 1800008, 10.1002/bies.201800008.
- Susmita Kaushik; Ana Maria Cuervo; The coming of age of chaperone-mediated autophagy. Nature Reviews Molecular Cell Biology 2018, 19, 365-381, 10.1038/s41580-018-0001-6.
- Yuchen Feng; Ding He; Zhiyuan Yao; Daniel J. Klionsky; The machinery of macroautophagy. Cell Research 2013, 24, 24-41, 10.1038/cr.2013.168.
- Damián Gatica; Vikramjit Lahiri; Daniel J. Klionsky; Cargo recognition and degradation by selective autophagy. Nature 2018, 20, 233-242, 10.1038/s41556-018-0037-z.
- Noboru Mizushima; Tamotsu Yoshimori; Yoshinori Ohsumi; The Role of Atg Proteins in Autophagosome Formation. Annual Review of Cell and Developmental Biology 2011, 27, 107-132, 10.1146/annurev-cellbio-092910-154005.
- Young Chul Kim; Kun-Liang Guan; mTOR: a pharmacologic target for autophagy regulation.. Journal of Clinical Investigation 2015, 125, 25-32, 10.1172/JCI73939.
- James H. Hurley; Lindsey N. Young; Mechanisms of Autophagy Initiation.. Annual Review of Biochemistry 2017, 86, 225-244, 10.1146/annurev-biochem-061516-044820.
- Chloe Burman; Nicholas T. Ktistakis; Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Letters 2010, 584, 1302-1312, 10.1016/j.febslet.2010.01.011.
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701.
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252.
- Hilla Weidberg; Elena Shvets; Tomer Shpilka; Frida Shimron; Vera Shinder; Zvulun Elazar; LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. The EMBO Journal 2010, 29, 1792-1802, 10.1038/emboj.2010.74.
- Daniel J. Klionsky; Kotb Abdelmohsen; Akihisa Abe; Joynal Abedin; Hagai Abeliovich; Abraham Acevedo Arozena; Hiroaki Adachi; Christopher M. Adams; Peter D Adams; Khosrow Adeli; et al.Peter J AdhihettySharon G AdlerGalila AgamRajesh AgarwalManish K AghiMaria AgnelloPatrizia AgostinisPatricia V AguilarJulio Aguirre-GhisoEdoardo M AiroldiSlimane Ait-Si-AliTakahiko AkematsuEmmanuel T AkporiayeMohamed Al-RubeaiGuillermo M. AlbaicetaChris AlbaneseDiego AlbaniMatthew L AlbertJesús AldudoHana AlgülMehrdad AlirezaeiIraide AllozaAlexandru AlmasanMaylin Almonte-BecerilEmad S AlnemriCovadonga AlonsoNihal Altan-BonnetDario C AltieriLydia Alvarez-ErvitiLydia Alvarez-ErvitiSandro AlvesG. AmadoroAtsuo AmanoConsuelo AmantiniSantiago AmbrosioIvano AmelioAmal O AmerMohamed AmessouAngelika AmonZhenyi AnFrank A AnaniaStig U AndersenUsha P. AndleyCatherine K AndreadiNathalie Andrieu-AbadieAlberto AnelDavid K AnnShailendra Anoopkumar-DukieManuela AntonioliHiroshi AokiNadezda ApostolovaSaveria AquilaKatia AquilanoKoichi ArakiEli AramaAgustín ArandaJun ArayaAlexandre ArcaroEsperanza AriasHirokazu Arimoto2)Aileen R AriosaJane ArmstrongThierry ArnouldIvica ArsovKatsuhiko AsanumaValerie AskanasÉric AsselinRyuichiro AtarashiSally S AthertonJulie D. AtkinLaura D AttardiPatrick AubergerGeorg AuburgerLaure AurelianRiccardo AutelliLaura AvaglianoMaria Laura AvantaggiatiLimor AvrahamiSuresh AwaleNeelam AzadTiziana BachettiJonathan M BackerDong-Hun BaeJae-Sung BaeOk-Nam BaeSoo Han BaeEric H BaehreckeSeung-Hoon BaekStephen BaghdiguianAgnieszka Bagniewska–ZadwornaHua BaiJie BaiXue-Yuan BaiYannick BaillyKithiganahalli Narayanaswamy BalajiWalter BalduiniAndrea BallabioRena BalzanRajkumar BanerjeeGabor BánhegyiHaijun BaoBenoit BarbeauMaria D BarrachinaEsther BarreiroBonnie BartelAlberto BartoloméDiane C BasshamMaria Teresa BassiRobert C. BastAlakananda BasuMaria Teresa BatistaHenri BatokoMaurizio BattinoKyle BauckmanBradley L BaumgarnerK Ulrich BayerRupert C.L. BealeJean-Francois BeaulieuGeorge R. BeckChristoph BeckerJ David BeckhamPierre-Andre BedardPatrick J BednarskiThomas J BegleyChristian BehlChristian BehrendsGeorg Mn BehrensKevin E BehrnsEloy BejaranoAmine BelaidFrancesca BelleudiGiovanni BenardGuy BerchemDaniele BergamaschiMatteo BergamiBen BerkhoutLaura BerliocchiAmélie BernardMonique BernardFrancesca BernassolaAnne BertolottiAmanda S BessSébastien BesteiroSaverio BettuzziSavita BhallaShalmoli BhattacharyyaSujit K BhutiaCaroline BiagoschMichele Wolfe BianchiMartine Biard-PiechaczykViktor BillesCláudia BincolettoBaris BingolSara W BirdMarc BitounIvana BjedovCraig BlackstoneLionel BlancGuillermo A BlancoHeidi Kiil BlomhoffEmilio Boada-RomeroStefan BöcklerMarianne BoesKathleen Boesze-BattagliaLawrence H BoiseAlessandra BolinoAndrea BomanPaolo BonaldoMatteo BordiJurgen BoschLuis M BotanaJoëlle BottiGerman BouMarina BoucheMarion BouchecareilhMarie-Josée BoucherMichael E BoultonSebastien G. BouretPatricia BoyaMichaël Boyer-GuittautPeter V. BozhkovNathan BradyVania Mm BragaClaudio BrancoliniGerhard H. BrausJosé M. Bravo-San PedroLisa A BrennanEmery H BresnickPatrick BrestDave BridgesMarie-Agnes BringerMarisa BriniGlauber C BritoBertha BrodinPaul S BrookesEric J BrownKaren BrownHal E BroxmeyerAlain BruhatPatrícia Chakur BrumJohn H BrumellNicola Brunetti-PierriRobert J. Bryson-RichardsonShilpa BuchAlastair M BuchanHikmet BudakDmitry V BulavinScott J BultmanGeert BultynckVladimir BumbasirevicYan BurelleRobert E BurkeMargit BurmeisterPeter BütikoferLaura CaberlottoKen CadwellMonika CahovaNgsheng CaiJingjing CaiQian CaiS. CalatayudNadine CamougrandMichelangelo CampanellaGrant R. CampbellMatthew CampbellSilvia CampelloRobin CandauIsabella CaniggiaLavinia CantoniLizhi CaoAllan B CaplanMichele CaragliaClaudio CardinaliS.M. CardosoJennifer S CarewLaura A CarletonCathleen R CarlinSilvia CarloniSven R. CarlssonDidac Carmona-GutierrezLeticia Am CarneiroOliana CarnevaliS CarraAlice CarrierB. CarrollCaty CasasJosefina CasasGiuliana CassinelliPerrine CastetsSusana Castro-ObregónGabriella CavalliniIsabella CeccheriniF CecconiArthur I CederbaumValentín CeñaSimone CenciClaudia CerellaDavide CerviaSilvia CetrulloHassan ChaachouayHan-Jung ChaeAndrei S. ChaginChee-Yin ChaiGopal ChakrabartiGeorgios ChamilosEdmond Yw ChanMatthew Tak Vai ChanDhyan ChandraPallavi ChandraMing-Shi ChangRaymond Chuen-Chung ChangTa Yuan ChangJohn C ChathamSaurabh ChatterjeeSantosh ChauhanYongsheng CheMichael E. CheethamRajkumar CheluvappaChun-Jung ChenGang ChenGuang-Chao ChenGuoqiang ChenHongzhuan ChenJeff W ChenJian-Kang ChenMin ChenMingzhou ChenPeiwen ChenQi ChenQuan ChenShang-Der ChenSi ChenSteve S-L ChenWei-Jung ChenWen Qiang ChenWenli ChenXiangmei ChenYau-Hung ChenYe-Guang ChenYin Chen Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1-222, 10.1080/15548627.2015.1100356.
- Yoshinori Takahashi; Haiyan He; Zhenyuan Tang; Tatsuya Hattori; Ying Liu; Megan M. Young; Jacob M. Serfass; Longgui Chen; Melat Gebru; Chong Chen; et al.Carson A. WillsJennifer M. AtkinsonHan ChenThomas AbrahamHong-Gang Wang An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure.. Nature Communications 2018, 9, 2855, 10.1038/s41467-018-05254-w.
- Shuhei Nakamura; Tamotsu Yoshimori; New insights into autophagosome–lysosome fusion. Journal of Cell Science 2017, 130, 1209-1216, 10.1242/jcs.196352.
- S. Pankiv; T. H. Clausen; Trond Lamark; A. Brech; Jack‐Ansgar Bruun1; H. Outzen; A. Overvatn; Geir Bjørkøy; Terje Johansen; p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. Journal of Biological Chemistry 2007, 282, 24131-24145, 10.1074/jbc.m702824200.
- Serhiy Pankiv; Endalkachew A. Alemu; Andreas Brech; Jack‐Ansgar Bruun1; Trond Lamark; Aud Øvervatn; Geir Bjørkøy; Terje Johansen; FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end–directed vesicle transport. Journal of Cell Biology 2010, 188, 253-269, 10.1083/jcb.200907015.
- Shunsuke Kimura; Takeshi Noda; Tamotsu Yoshimori; Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes.. Cell Structure and Function 2008, 33, 109-122, 10.1247/csf.08005.
- Peidu Jiang; Taki Nishimura; Yuriko Sakamaki; Eisuke Itakura; Tomohisa Hatta; Tohru Natsume; Noboru Mizushima; The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Molecular Biology of the Cell 2014, 25, 1327-1337, 10.1091/mbc.E13-08-0447.
- Ruslan Medzhitov; Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771-776, 10.1016/j.cell.2010.03.006.
- Elzbieta Kolaczkowska; Paul Kubes; Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology 2013, 13, 159-175, 10.1038/nri3399.
- Aimee M Decathelineau; Peter M Henson; The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays in Biochemistry 2003, 39, 105-117, 10.1042/bse0390105.
- Charles N. Serhan; Sue D. Brain; Christopher D. Buckley; Derek W. Gilroy; Chris Haslett; Luke A. J. O’Neill; Mauro Perretti; Adriano G. Rossi; John L. Wallace; Resolution of inflammation: state of the art, definitions and terms. The FASEB Journal 2006, 21, 672271, 10.1096/fj.06-7227com.
- Satoshi Watanabe; Michael Alexander; Alexander V. Misharin; G. R. Scott Budinger; The role of macrophages in the resolution of inflammation. Journal of Clinical Investigation 2019, 129, 2619-2628, 10.1172/JCI124615.
- Michelle Amantéa Sugimoto; Juliana Priscila Vago; Mauro Perretti; Mauro M. Teixeira; Mediators of the Resolution of the Inflammatory Response. Trends in Immunology 2019, 40, 212-227, 10.1016/j.it.2019.01.007.
- Almudena Ortega-Gomez; Mauro Perretti; Oliver Soehnlein; Resolution of inflammation: an integrated view. EMBO Molecular Medicine 2013, 5, 661-674, 10.1002/emmm.201202382.
- Charles N. Serhan; Bruce D. Levy; Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. Journal of Clinical Investigation 2018, 128, 2657-2669, 10.1172/jci97943.
- Bruce D. Levy; Clary B Clish; Birgitta Schmidt; Karsten Gronert; Charles N. Serhan; Lipid mediator class switching during acute inflammation: signals in resolution. Nature Immunology 2001, 2, 612-619, 10.1038/89759.
- Antonio Recchiuti; Charles N. Serhan; Pro-Resolving Lipid Mediators (SPMs) and Their Actions in Regulating miRNA in Novel Resolution Circuits in Inflammation. Frontiers in Immunology 2012, 3, 298, 10.3389/fimmu.2012.00298.
- C. N. Serhan; M. Hamberg; B. Samuelsson; Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes.. Proceedings of the National Academy of Sciences 1984, 81, 5335-5339, 10.1073/pnas.81.17.5335.
- Serhan, C.N.; Sheppard, K.A. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Investig. 1990, 85, 772–780.
- Romano, M.; Chen, X.S.; Takahashi, Y.; Yamamoto, S.; Funk, C.D.; Serhan, C.N. Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem. J. 1993, 296, 127–133.
- Edenius, C.; Kumlin, M.; Björk, T.; Änggård, A.; Lindgren, J.Å. Lipoxin formation in human nasal polyps and bronchial tissue. FEBS Lett. 1990, 272, 25–28.
- Levy, B.D.; Romano, M.; Chapman, H.A.; Reilly, J.J.; Drazen, J.; Serhan, C.N. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J. Clin. Investig. 1993, 92, 1572–1579.
- Joan Claria; C. N. Serhan; Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions.. Proceedings of the National Academy of Sciences 1995, 92, 9475-9479, 10.1073/pnas.92.21.9475.
- Charles N. Serhan; Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. The FASEB Journal 2017, 31, 1273-1288, 10.1096/fj.201601222r.
- Charles N. Serhan; Clary B Clish; Jessica Brannon; Sean P. Colgan; Nan Chiang; Karsten Gronert; Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2–Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. Journal of Experimental Medicine 2000, 192, 1197-1204, 10.1084/jem.192.8.1197.
- Jesmond Dalli; Nan Chiang; Charles N. Serhan; Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nature Medicine 2015, 21, 1071-1075, 10.1038/nm.3911.
- Charles N. Serhan; Jesmond Dalli; Romain A. Colas; Jeremy W. Winkler; Nan Chiang; Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2015, 1851, 397-413, 10.1016/j.bbalip.2014.08.006.
- Charles N. Serhan; Rong Yang; Kimberly Martinod; Kie Kasuga; Padmini S. Pillai; Timothy F. Porter; Sungwhan F. Oh; Matthew Spite; Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. Journal of Experimental Medicine 2008, 206, 15-23, 10.1084/jem.20081880.
- Jesmond Dalli; Sesquile Ramon; Paul C. Norris; Romain A. Colas; Charles N. Serhan; Novel proresolving and tissue‐regenerative resolvin and protectin sulfido‐conjugated pathways. The FASEB Journal 2015, 29, 2120-2136, 10.1096/fj.14-268441.
- Jesmond Dalli; Iliyan Vlasakov; Ian R. Riley; Ana R. Rodriguez; Bernd W. Spur; Nicos A. Petasis; Nan Chiang; Charles N. Serhan; Maresin conjugates in tissue regeneration biosynthesis enzymes in human macrophages. Proceedings of the National Academy of Sciences 2016, 113, 12232-12237, 10.1073/pnas.1607003113.
- Jesmond Dalli; Romain A. Colas; Charles N. Serhan; Novel n-3 Immunoresolvents: Structures and Actions. Scientific Reports 2013, 3, srep01940, 10.1038/srep01940.
- Nan Chiang; Charles N. Serhan; Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors.. Molecular Aspects of Medicine 2017, 58, 114-129, 10.1016/j.mam.2017.03.005.
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665.
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722.
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461.
- S Fiore; S W Ryeom; P F Weller; C N Serhan; Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils.. Journal of Biological Chemistry 1992, 267, 16168–16176.
- Mario Romano; Irene Recchia; Antonio Recchiuti; Lipoxin Receptors. The Scientific World Journal 2007, 7, 1393-1412, 10.1100/tsw.2007.186.
- Mario Romano; Sara Patruno; Antonella Pomilio; Antonio Recchiuti; Proresolving Lipid Mediators and Receptors in Stem Cell Biology: Concise Review. STEM CELLS Translational Medicine 2019, 8, 992-998, 10.1002/sctm.19-0078.
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Fredman, G.; Serhan, C.N. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 2012, 180, 2018–2027.
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528.
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.-Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Potent Immunoresolvents. Chem. Biol. 2013, 20, 188–201.
- Arnardottir, H.H.; Dalli, J.; Norling, L.V.; Colas, R.A.; Perretti, M.; Serhan, C.N. Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation. J. Immunol. 2016, 197, 2362–2368.
- Olivier Corminboeuf; Xavier Leroy; FPR2/ALXR Agonists and the Resolution of Inflammation. Journal of Medicinal Chemistry 2014, 58, 537-559, 10.1021/jm501051x.
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-Inflammatory Role of the Murine Formyl-Peptide Receptor 2: Ligand-Specific Effects on Leukocyte Responses and Experimental Inflammation. J. Immunol. 2010, 184, 2611–2619.
- Devchand, P.R.; Arita, M.; Hong, S.; Bannenberg, G.; Moussignac, R.; Gronert, K.; Serhan, C.N. Human ALX receptor regulates neutrophil recruitment in transgenic mice: Roles in inflammation and host defense. FASEB J. 2003, 17, 652–659.
- Simiele, F.; Recchiuti, A.; Mattoscio, D.; De Luca, A.; Cianci, E.; Franchi, S.; Gatta, V.; Parolari, A.; Werba, J.P.; Camera, M.; et al. Transcriptional regulation of the human FPR2/ALX gene: Evidence of a heritable genetic variant that impairs promoter activity. FASEB J. 2012, 26, 1323–1333.
- Zhang, H.; Lu, Y.; Sun, G.; Teng, F.; Luo, N.; Jiang, J.; Wen, A. The common promoter polymorphism rs11666254 downregulates FPR2/ALX expression and increases risk of sepsis in patients with severe trauma. Crit. Care 2017, 21, 171.
- Nan Chiang; Jesmond Dalli; Romain A. Colas; Charles N. Serhan; Identification of resolvin D2 receptor mediating resolution of infections and organ protection. Journal of Experimental Medicine 2015, 212, 1203-1217, 10.1084/jem.20150225.
- Nan Chiang; Xavier De La Rosa; Stephania Libreros; Charles N. Serhan; Novel Resolvin D2 Receptor Axis in Infectious Inflammation.. The Journal of Immunology 2016, 198, 842-851, 10.4049/jimmunol.1601650.
- Makoto Arita; Taisuke Ohira; Yee-Ping Sun; Siva Elangovan; Nan Chiang; Charles N. Serhan; Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation.. The Journal of Immunology 2007, 178, 3912-3917, 10.4049/jimmunol.178.6.3912.
- Nan Chiang; Stephania Libreros; Paul C. Norris; Xavier De La Rosa; Charles N. Serhan; Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. Journal of Clinical Investigation 2019, 129, 5294-5311, 10.1172/jci129448.
- Sangsu Bang; Ya-Kai Xie; Zhi-Jun Zhang; Zilong Wang; Zhen-Zhong Xu; Ru-Rong Ji; GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. Journal of Clinical Investigation 2018, 128, 3568-3582, 10.1172/jci99888.
- Magdalena B. Flak; Duco S. Koenis; Agua Sobrino; James Smith; Kimberly Pistorius; Francesco Palmas; Jesmond Dalli; GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. Journal of Clinical Investigation 2019, 130, 359-373, 10.1172/jci131609.
- Zenglin Liao; Jiajia Dong; Wei Wu; Ting Yang; Tao Wang; Lingli Guo; Lei Chen; Dan Xu; Fu-Qiang Wen; Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respiratory Research 2012, 13, 110-110, 10.1186/1465-9921-13-110.
- David Keinan; Noel J. Leigh; Joel W. Nelson; Laura De Oleo; Olga J. Baker; Understanding Resolvin Signaling Pathways to Improve Oral Health. International Journal of Molecular Sciences 2013, 14, 5501-5518, 10.3390/ijms14035501.
- Antonio Recchiuti; Sriram Krishnamoorthy; Gabrielle Fredman; Nan Chiang; Charles N. Serhan; MicroRNAs in resolution of acute inflammation: identification of novel resolvin Dl‐miRNA circuits. The FASEB Journal 2010, 25, 544-560, 10.1096/fj.10-169599.
- Charles N. Serhan; Nan Chiang; Jesmond Dalli; The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution.. Seminars in Immunology 2015, 27, 200-215, 10.1016/j.smim.2015.03.004.
- Matthew D. Keller; Victor J. Torres; Ken Cadwell; Autophagy and microbial pathogenesis. Cell Death & Differentiation 2020, 27, 872-886, 10.1038/s41418-019-0481-8.
- Do Hoon Kwon; Hyun Kyu Song; A Structural View of Xenophagy, a Battle between Host and Microbes. Molecules and Cells 2018, 41, 27-34, 10.14348/molcells.2018.2274.
- Bradlee L. Heckmann; Emilio Boada-Romero; Larissa D. Cunha; Joelle Magné; Douglas R. Green; LC3-Associated Phagocytosis and Inflammation.. Journal of Molecular Biology 2017, 429, 3561-3576, 10.1016/j.jmb.2017.08.012.
- Yuan He; Hideki Hara; Gabriel Núñez; Mechanism and Regulation of NLRP3 Inflammasome Activation.. Trends in Biochemical Sciences 2016, 41, 1012-1021, 10.1016/j.tibs.2016.09.002.
- Qiuyun Liu; Danyan Zhang; Diyu Hu; Xiangmei Zhou; Yang Zhou; The role of mitochondria in NLRP3 inflammasome activation. Molecular Immunology 2018, 103, 115-124, 10.1016/j.molimm.2018.09.010.
- Anne Hamacher-Brady; Nathan R Brady; Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cellular and Molecular Life Sciences 2015, 73, 775-795, 10.1007/s00018-015-2087-8.
- Qian Sun; Jie Fan; Timothy R. Billiar; Melanie J. Scott; Sun Qian; Inflammasome and Autophagy Regulation: A Two-way Street. Molecular Medicine 2017, 23, 188-195, 10.2119/molmed.2017.00077.
- Tatsuya Saitoh; Naonobu Fujita; Myoung Ho Jang; Satoshi Uematsu; Bo-Gie Yang; Takashi Satoh; Hiroko Omori; Takeshi Noda; Naoki Yamamoto; Masaaki Komatsu; et al.Keiji TanakaTaro KawaiTohru TsujimuraOsamu TakeuchiTamotsu YoshimoriShizuo Akira Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264-268, 10.1038/nature07383.
- Yu Tian; Ming-Li Wang; Jun Zhao; Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132, 10.3390/v11020132.
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T.; et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host Microbe 2014, 15, 228–238.
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846.
- Konno, H.; Konno, K.; Barber, G.N. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155, 688–698.
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775.
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.-Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055
- Ting Liu; Lingyun Zhang; Donghyun Joo; Shao-Cong Sun; NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy 2017, 2, 17023, 10.1038/sigtrans.2017.23.
- Alain Israel; The IKK Complex, a Central Regulator of NF- B Activation. Cold Spring Harbor Perspectives in Biology 2009, 2, a000158-a000158, 10.1101/cshperspect.a000158.
- Yuri Shibata; Masaaki Oyama; Hiroko Kozuka-Hata; Xiao Han; Yuetsu Tanaka; Jin Gohda; Jun-Ichiro Inoue; p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nature Communications 2012, 3, 1061, 10.1038/ncomms2068.
- Kunpeng Liu; Lei Zhang; Qiang Zhao; Zhiyao Zhao; Feng Zhi; Yunfei Qin; Jun Cui; SKP2 attenuates NF-κB signaling by mediating IKKβ degradation through autophagy. Journal of Molecular Cell Biology 2018, 10, 205-215, 10.1093/jmcb/mjy012.
- Yu Zhang; Xian Guo; Wanyao Yan; Yan Chen; Mengxiang Ke; Cheng Cheng; Xiuqin Zhu; Weili Xue; Qiaoqiao Zhou; Ling Zheng; et al.Shun WangBin WuXinran LiuLiang MaLianqi HuangKun Huang ANGPTL8 negatively regulates NF-κB activation by facilitating selective autophagic degradation of IKKγ.. Nature Communications 2017, 8, 2164, 10.1038/s41467-017-02355-w.
- Nina Germic; Ziva Frangez; Shida Yousefi; Hans-Uwe Simon; Regulation of the innate immune system by autophagy: neutrophils, eosinophils, mast cells, NK cells. Cell Death & Differentiation 2019, 26, 703-714, 10.1038/s41418-019-0295-8.
- S Rožman; Shida Yousefi; K Oberson; Thomas Kaufmann; Charaf Benarafa; Hans-Uwe Simon; The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death & Differentiation 2014, 22, 445-456, 10.1038/cdd.2014.169.
- Hiroshi Itoh; Hidemasa Matsuo; Naoko Kitamura; Sho Yamamoto; Takeshi Higuchi; Hiromu Takematsu; Yasuhiko Kamikubo; Tadakazu Kondo; Kouhei Yamashita; Masataka Sasada; et al.Akifumi Takaori-KondoSouichi Adachi Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. Journal of Leukocyte Biology 2015, 98, 107-117, 10.1189/jlb.4a0813-422rrr.
- Quinten Remijsen; Tom Vandenberghe; Ellen Wirawan; Bob Asselbergh; Eef Parthoens; Riet De Rycke; Samuel Noppen; Michel Delforge; Jean Willems; Peter Vandenabeele; et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Research 2010, 21, 290-304, 10.1038/cr.2010.150.
- Abhisek Bhattacharya; Qin Wei; Jin Na Shin; ElMoataz Abdel Fattah; Diana L. Bonilla; Qian Xiang; N. Tony Eissa; Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Reports 2015, 12, 1731-1739, 10.1016/j.celrep.2015.08.019.
- Nina Germic; Ziva Frangez; Shida Yousefi; Hans-Uwe Simon; Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death & Differentiation 2019, 26, 715-727, 10.1038/s41418-019-0297-6.
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905.
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1–induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012, 119, 4527–4531
- Ming-Yue Wu; Jia-Hong Lu; Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70, 10.3390/cells9010070.
- Crotzer, V.L.; Blum, J.S. Autophagy and adaptive immunity: Autophagy and immunity. Immunology 2010, 131, 9–17.
- Münz, C. Autophagy in immunity. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 172, pp. 67–85. ISBN 978-0-12-822021-4.
- Qingdong Guan; A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. Journal of Immunology Research 2019, 2019, 1-16, 10.1155/2019/7247238.
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211.
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604.
- Kara G. Lassen; Petric Kuballa; Kara L. Conway; Khushbu K. Patel; Christine E. Becker; Joanna M. Peloquin; Eduardo J. Villablanca; Jason M. Norman; Ta-Chiang Liu; Robert J. Heath; et al.Morgan L. BeckerLola FagbamiHeiko HornJohnathan MercerOmer H. YilmazJacob D. JaffeAlykhan F. ShamjiAtul K. BhanSteven A. CarrMark J. DalyHerbert W. VirginStuart L. SchreiberThaddeus S. StappenbeckRamnik J. Xavier Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proceedings of the National Academy of Sciences 2014, 111, 7741-7746, 10.1073/pnas.1407001111.
- Ken Cadwell; John Y. Liu; Sarah L. Brown; Hiroyuki Miyoshi; Joy Loh; Jochen K. Lennerz; Chieko Kishi; Wumesh Kc; Javier A. Carrero; Steven Hunt; et al.Christian D. StoneElizabeth M. BruntRamnik J. XavierBarry P. SleckmanEllen LiNoboru MizushimaThaddeus S. StappenbeckHerbert W. Virgin A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259-263, 10.1038/nature07416.
- Hong Zhang; Libo Zheng; Dermot P. B. McGovern; Ariel M. Hamill; Ryan Ichikawa; Yoshitake Kanazawa; Justin Luu; Kotaro Kumagai; Marianne Cilluffo; Masayuki Fukata; et al.Stephan R. TarganDavid M. UnderhillXiaolan ZhangDavid Q. Shih Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis.. The Journal of Immunology 2017, 198, 2133-2146, 10.4049/jimmunol.1601293.
- Alexandra C. Racanelli; Sarah Ann Kikkers; Augustine M.K. Choi; Suzanne M. Cloonan; Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221-232, 10.1080/15548627.2017.1389823.
- J.R. Riordan; Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Trends in Genetics 1989, 5, 363, 10.1016/0168-9525(89)90155-8.
- Anita Balázs; Marcus A. Mall; Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway.. Pediatric Pulmonology 2019, 54, S5-S12, 10.1002/ppul.24462.
- André M. Cantin; Minik Hartl; Michael W. Konstan; James F. Chmiel; Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. Journal of Cystic Fibrosis 2015, 14, 419-430, 10.1016/j.jcf.2015.03.003.
- Karp, C.L.; Flick, L.M.; Park, K.W.; Softic, S.; Greer, T.M.; Keledjian, R.; Yang, R.; Uddin, J.; Guggino, W.B.; Atabani, S.F.; et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat. Immunol. 2004, 5, 388–392.
- Mattoscio, D.; Evangelista, V.; De Cristofaro, R.; Recchiuti, A.; Pandolfi, A.; Di Silvestre, S.; Manarini, S.; Martelli, N.; Rocca, B.; Petrucci, G.; et al. Cystic fibrosis transmembrane conductance regulator (CFTR) expression in human platelets: Impact on mediators and mechanisms of the inflammatory response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3970–3980
- Alessandro Luciani; Valeria Rachela Villella; Speranza Esposito; Nicola Brunetti-Pierri; Diego L. Medina; Carmine Settembre; Manuela Gavina; Laura Pulze; Ida Giardino; Massimo Pettoello-Mantovani; et al.Maria D’ApolitoStefano GuidoEliezer MasliahBrian SpencerSonia QuaratinoValeria RaiaAndrea BallabioLuigi Maiuri Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature 2010, 12, 863-875, 10.1038/ncb2090.
- Rossana G. Iannitti; Valerio Napolioni; Vasilis Oikonomou; Antonella De Luca; Claudia Galosi; Marilena Pariano; Cristina Massi-Benedetti; Monica Borghi; Matteo Puccetti; Vincenzina Lucidi; et al.Carla ColomboErsilia FiscarelliCornelia Lass-FlörlFabio MajoLisa CarianiMaria RussoLuigi PorcaroGabriella RicciottiHelmut EllemunterLuigi RatclifFernando Maria De BenedictisVincenzo Nicola TalesaCharles A. DinarelloFrank L. Van De VeerdonkLuigina Romani IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nature Communications 2016, 7, 10791, 10.1038/ncomms10791.
- Matthew L. Mayer; Christoph J. Blohmke; Reza Falsafi; Chris D. Fjell; Laurence Madera; Stuart E. Turvey; Robert E. W. Hancock; Christopher D. Fjell; Rescue of Dysfunctional Autophagy Attenuates Hyperinflammatory Responses from Cystic Fibrosis Cells. The Journal of Immunology 2012, 190, 1227-1238, 10.4049/jimmunol.1201404.
- Valeria R. Villella; Speranza Esposito; Eleonora Ferrari; Romina Monzani; Antonella Tosco; Federica Rossin; Alice Castaldo; Marco Silano; Gian Luigi Marseglia; Luigina Romani; et al.Nikolai A. BarlevMauro PiacentiniValeria RaiaGuido KroemerLuigi Maiuri Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death & Disease 2019, 10, 258, 10.1038/s41419-019-1500-x.
- Kyle Caution; Alexander Pan; Kathrin Krause; Asmaa Badr; Kaitlin Hamilton; Anup Vaidya; Hawin Gosu; Kylene Daily; Shady Estfanous; Mikhail A. Gavrilin; et al.Mark E. DrewEstelle Cormet-BoyakaXi ChenDavid E. FrankhouserRalf BundschuhPearlly YanDuaa DakhlallahAmal O. Amer Methylomic correlates of autophagy activity in cystic fibrosis. Journal of Cystic Fibrosis 2019, 18, 491-500, 10.1016/j.jcf.2019.01.011.
- Mia F. Tazi; Duaa A. Dakhlallah; Kyle Caution; Madelyn M. Gerber; Sheng-Wei Chang; Hany Khalil; Benjamin T. Kopp; Amr E. Ahmed; Kathrin Krause; Ian Davis; et al.Clay MarshAmy E. Lovett-RackeLarry S. SchlesingerEstelle Cormet-BoyakaAmal O. Amer Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 2016, 12, 2026-2037, 10.1080/15548627.2016.1217370.
- Basant A. Abdulrahman; Arwa Abu Khweek; Anwari Akhter; Kyle Caution; Sheetal Kotrange; Dalia H.A. Abdelaziz; Christie Newland; Roberto Rosales-Reyes; Benjamin T. Kopp; Karen McCoy; et al.Richard MontioneLarry S. SchlesingerMikhail A. GavrilinMark D. WewersMiguel Ángel ValvanoAmal O. Amer Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011, 7, 1359-1370, 10.4161/auto.7.11.17660.
- Basant A. Abdulrahman; Arwa Abu Khweek; Anwari Akhter; Kyle Caution; Mia Tazi; Hoda Hassan; Yucheng Zhang; Patrick D. Rowland; Sankalp Malhotra; Famke Aeffner; et al.Ian C. DavisMiguel Ángel ValvanoAmal O. Amer Depletion of the Ubiquitin-binding Adaptor Molecule SQSTM1/p62 from Macrophages Harboring cftr ΔF508 Mutation Improves the Delivery of Burkholderia cenocepacia to the Autophagic Machinery*. Journal of Biological Chemistry 2012, 288, 2049-2058, 10.1074/jbc.M112.411728.
- Robert D. Junkins; Ann Shen; Kirill Rosen; Craig McCormick; Tong-Jun Lin; Autophagy Enhances Bacterial Clearance during P. aeruginosa Lung Infection. PLOS ONE 2013, 8, e72263, 10.1371/journal.pone.0072263.
- Maurizio Renna; Catherine Schaffner; Karen Brown; Shaobin Shang; Marcela Henao Tamayo; Krisztina Hegyi; Neil Grimsey; David Cusens; Sarah Coulter; Jason Cooper; et al.Anne R. BowdenSandra M. NewtonBeate KampmannJennifer HelmAndrew M JonesCharles S. HaworthRandall J. BasarabaMary Ann DeGrooteDiane OrdwayDavid C. RubinszteinR. Andres Floto Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. Journal of Clinical Investigation 2011, 121, 3554-3563, 10.1172/jci46095.
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074.
- Tosco, A.; De Gregorio, F.; Esposito, S.; De Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; Di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393.
- Shaoyi Zhang; Gautier Stoll; José M. Bravo-San Pedro; Valentina Sica; Allan Sauvat; Florine Obrist; Oliver Kepp; Yousheng Li; Luigi Maiuri; Naoufal Zamzami; et al.Guido Kroemer Evaluation of autophagy inducers in epithelial cells carrying the ΔF508 mutation of the cystic fibrosis transmembrane conductance regulator CFTR.. Cell Death & Disease 2018, 9, 191, 10.1038/s41419-017-0235-9.
- Manish Bodas; Neeraj Vij; Adapting Proteostasis and Autophagy for Controlling the Pathogenesis of Cystic Fibrosis Lung Disease. Frontiers in Pharmacology 2019, 10, 20, 10.3389/fphar.2019.00020.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Shikha Satendra Singh; Somya Vats; Amelia Yi-Qian Chia; Tuan Zea Tan; Shuo Deng; Mei Shan Ong; Frank Arfuso; Celestial T. Yap; Boon Cher Goh; Gautam Sethi; et al.Ruby Yun-Ju HuangHan-Ming ShenRavi ManjithayaAlan Prem Kumar Dual role of autophagy in hallmarks of cancer. Oncogene 2017, 37, 1142-1158, 10.1038/s41388-017-0046-6.
- Mattoscio, D.; Casadio, C.; Miccolo, C.; Maffini, F.; Raimondi, A.; Tacchetti, C.; Gheit, T.; Tagliabue, M.; Galimberti, V.E.; De Lorenzi, F.; et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017, 13, e1006262.
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775.
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.; Donnelly, C.R.; Luo, X.; Heath, B.R.; Zhao, X.; Bellile, E.; Hu, H.; Chen, H.; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin. Cancer Res. 2018, 24, 4242–4255.
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652.
- Shenghong Yang; Yu Imamura; Russell W. Jenkins; Israel Cañadas; Shunsuke Kitajima; Amir R Aref; Arthur L Brannon; Eiji Oki; Adam Castoreno; Zehua Zhu; et al.Tran C ThaiJacob ReibelZhirong QianShuji OginoKwok K. WongHideo BabaAlec C. KimmelmanMarina Pasca Di MaglianoDavid A. Barbie Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation.. Cancer Immunology Research 2016, 4, 520-530, 10.1158/2326-6066.CIR-15-0235.
- Robin Mathew; Sinan Khor; Sean R. Hackett; Joshua D. Rabinowitz; David H. Perlman; Eileen White; Functional Role of Autophagy-Mediated Proteome Remodeling in Cell Survival Signaling and Innate Immunity. Molecular Cell 2014, 55, 916-930, 10.1016/j.molcel.2014.07.019.
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056.
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577
- Sang‐Man Jin; Hye Won Jang; Seo Young Sohn; Na Kyung Kim; Ji Young Joung; Yoon Young Cho; Sun Wook Kim; Jae Hoon Chung; Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine 2013, 45, 256-262, 10.1007/s12020-013-9997-8.
- Elisa Isopi; Alberto Granzotto; Carlo Corona; Manuela Bomba; Domenico Ciavardelli; Michele Curcio; Lorella M.T. Canzoniero; Riccardo Navarra; Rossano Lattanzio; Mauro Piantelli; et al.Stefano L. Sensi Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer's disease without reducing amyloid and tau pathology. Neurobiology of Disease 2015, 81, 214-224, 10.1016/j.nbd.2014.11.013.
- Frank L. Heppner; Richard M. Ransohoff; Burkhard Becher; Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience 2015, 16, 358-372, 10.1038/nrn3880.
- Rongbin Zhou; Amir S. Yazdi; Philippe Menu; Jurg Tschopp; Erratum: A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 475, 122-122, 10.1038/nature10156.
- Douglas R. Green; Lorenzo Galluzzi; Guido Kroemer; Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109-1112, 10.1126/science.1201940.
- Mehmet Bostanciklioğlu; Mehmet Bostanciklioğlu; An update on the interactions between Alzheimer's disease, autophagy and inflammation.. Gene 2019, 705, 157-166, 10.1016/j.gene.2019.04.040.
- Evandro Fei Fang; Yujun Hou; Konstantinos Palikaras; Bryan A. Adriaanse; Jesse S. Kerr; Beimeng Yang; Sofie Lautrup; Mahdi Hasan-Olive; Domenica Caponio; Xiuli Dan; et al.Paula RocktäschelDeborah L. CroteauMansour AkbariNigel H. GreigTormod FladbyHilde NilsenM. Zameel CaderMark P. MattsonNektarios TavernarakisVilhelm A. Bohr Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nature Neuroscience 2019, 22, 401-412, 10.1038/s41593-018-0332-9.
- Patricia Prieto; Cesar Eduardo Rosales-Mendoza; Verónica Terrón; Victor Toledano; Antonio Cuadrado; Eduardo López-Collazo; G. Bannenberg; Paloma Martin-Sanz; María Fernández-Velasco; Lisardo Boscá; et al. Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy 2015, 11, 1729-1744, 10.1080/15548627.2015.1078958.
- Huaizheng Liu; Kefu Zhou; Liangkan Liao; Tianyi Zhang; Mingshi Yang; Chuanzheng Sun; Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respiratory Research 2018, 19, 243, 10.1186/s12931-018-0937-2.
- Juan Xiao; Xueping Feng; Xiao-Ying Huang; Zhongshi Huang; Yanqiang Huang; Chaogan Li; Genliang Li; Song Nong; Ruoshi Wu; Yongzhi Huang; et al.Xi-Dai Long Spautin-1 Ameliorates Acute Pancreatitis via Inhibiting Impaired Autophagy and Alleviating Calcium Overload. Molecular Medicine 2016, 22, 643-652, 10.2119/molmed.2016.00034.
- Bingbing Wang; Cui Hu; Yongyu Mei; Junjun Bao; Shaozhen Ding; Xiaochang Liu; Qiao Mei; Jianming Xu; Resolvin D1 Resolve Inflammation in Experimental Acute Pancreatitis by Restoring Autophagic Flux. Digestive Diseases and Sciences 2018, 63, 3359-3366, 10.1007/s10620-018-5191-4.
- Zhang, J.; Wang, M.; Ding, W.; Zhao, M.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; Li, D.; Liu, J.; et al.et al. Resolvin E1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting AKT/mTOR signaling. Biochem. Pharmacol. 2020, 180, 114188, 10.1016/j.bcp.2020.114188.
- Emma Börgeson; Andrew M.F. Johnson; Yun Sok Lee; Andreas Till; Gulam Hussain Syed; Syed Tasadaque Ali-Shah; Patrick J. Guiry; Jesmond Dalli; Romain A. Colas; Charles N. Serhan; et al.Kumar SharmaCatherine Godson Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease.. Cell Metabolism 2015, 22, 125-137, 10.1016/j.cmet.2015.05.003.
- Rinke Stienstra; Yulia Haim; Yael Riahi; Mihai Netea; Assaf Rudich; Gil Leibowitz; Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 2014, 57, 1505-1516, 10.1007/s00125-014-3255-3.
- Laura M Laiglesia; S. Lorente-Cebrián; Miguel López-Yoldi; Raquel Lanas; Neira Sáinz; José Alfredo Martínez; María Jesús Moreno-Aliaga; Maresin 1 inhibits TNF-alpha-induced lipolysis and autophagy in 3T3-L1 adipocytes. Journal of Cellular Physiology 2017, 233, 2238-2246, 10.1002/jcp.26096.
- Li Du; Yucheng Li; Weifeng Liu; Maresin 1 regulates autophagy and inflammation in human periodontal ligament cells through glycogen synthase kinase–3β/β-catenin pathway under inflammatory conditions. Archives of Oral Biology 2018, 87, 242-247, 10.1016/j.archoralbio.2017.12.023.
- Ping Yin; Xu Wang; Shuang Wang; Yafen Wei; Jiachun Feng; Mingqin Zhu; Maresin 1 Improves Cognitive Decline and Ameliorates Inflammation in a Mouse Model of Alzheimer’s Disease. Frontiers in Cellular Neuroscience 2019, 13, 466, 10.3389/fncel.2019.00466.
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 960, pp. 277–304. ISBN 978-3-319-48380-1.
- Cristina López-Vicario; José Alcaraz-Quiles; Verónica García-Alonso; Bibiana Rius; Sung H. Hwang; Esther Titos; Aritz Lopategi; Bruce D. Hammock; Vicente Arroyo; Joan Claria; et al. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proceedings of the National Academy of Sciences 2014, 112, 536-541, 10.1073/pnas.1422590112.

