 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlota Leonardo-Sousa | -- | 5859 | 2022-04-14 18:11:40 | | | |
| 2 | Vivi Li | Meta information modification | 5859 | 2022-04-15 05:47:14 | | | | |
| 3 | Vivi Li | Meta information modification | 5859 | 2022-04-18 08:46:05 | | |
Video Upload Options
Proteasome inhibitors have shown relevant clinical activity in several hematological malignancies, namely in multiple myeloma and mantle cell lymphoma, improving patient outcomes such as survival and quality of life, when compared with other therapies. However, initial response to the therapy is a challenge as most patients show an innate resistance to proteasome inhibitors, and those that respond to the therapy usually develop late relapses suggesting the development of acquired resistance. The mechanisms of resistance to proteasome inhibition are still controversial and scarce in the literature.
1. Introduction
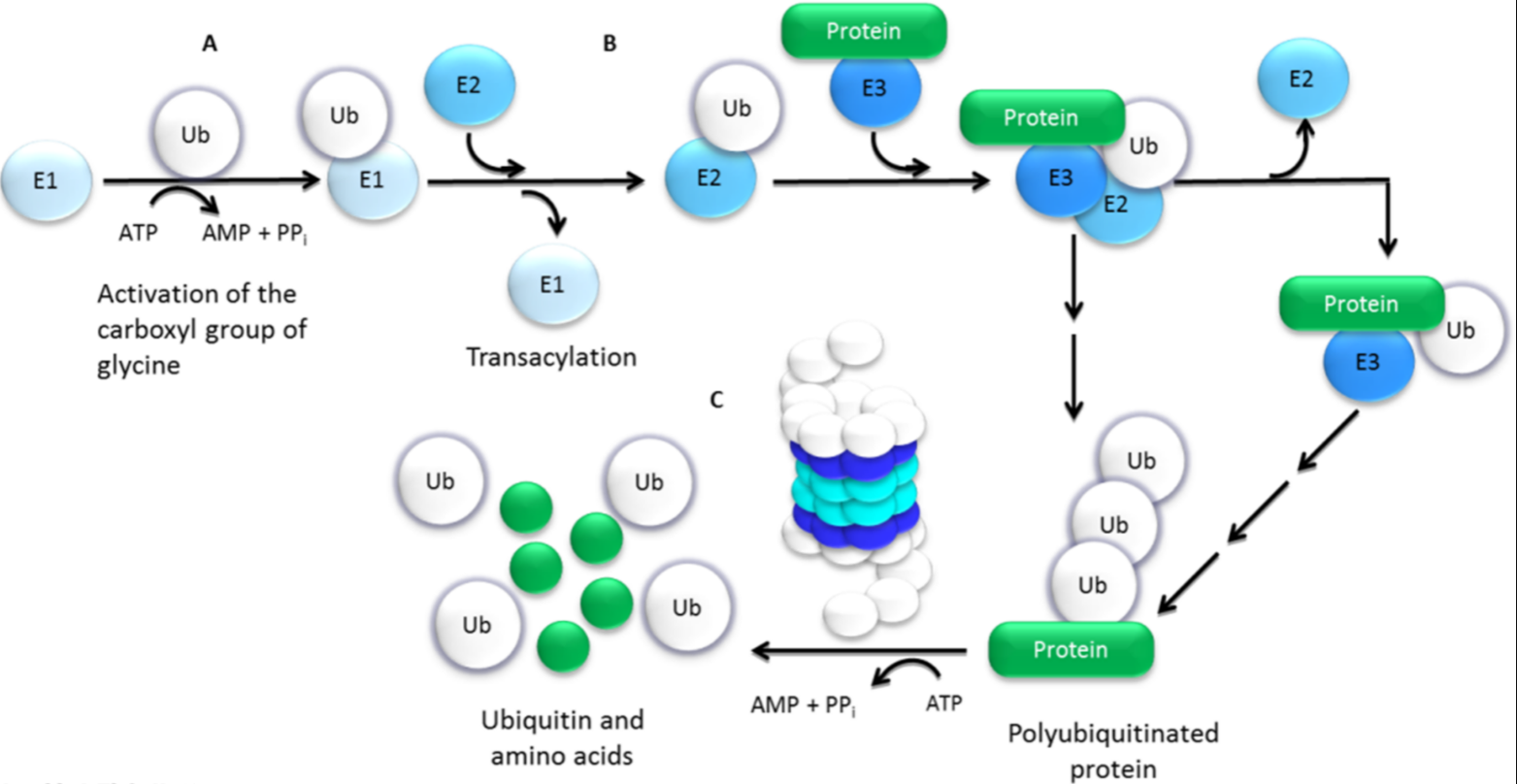
2. Ubiquitin–Proteasome Pathway (UPP)
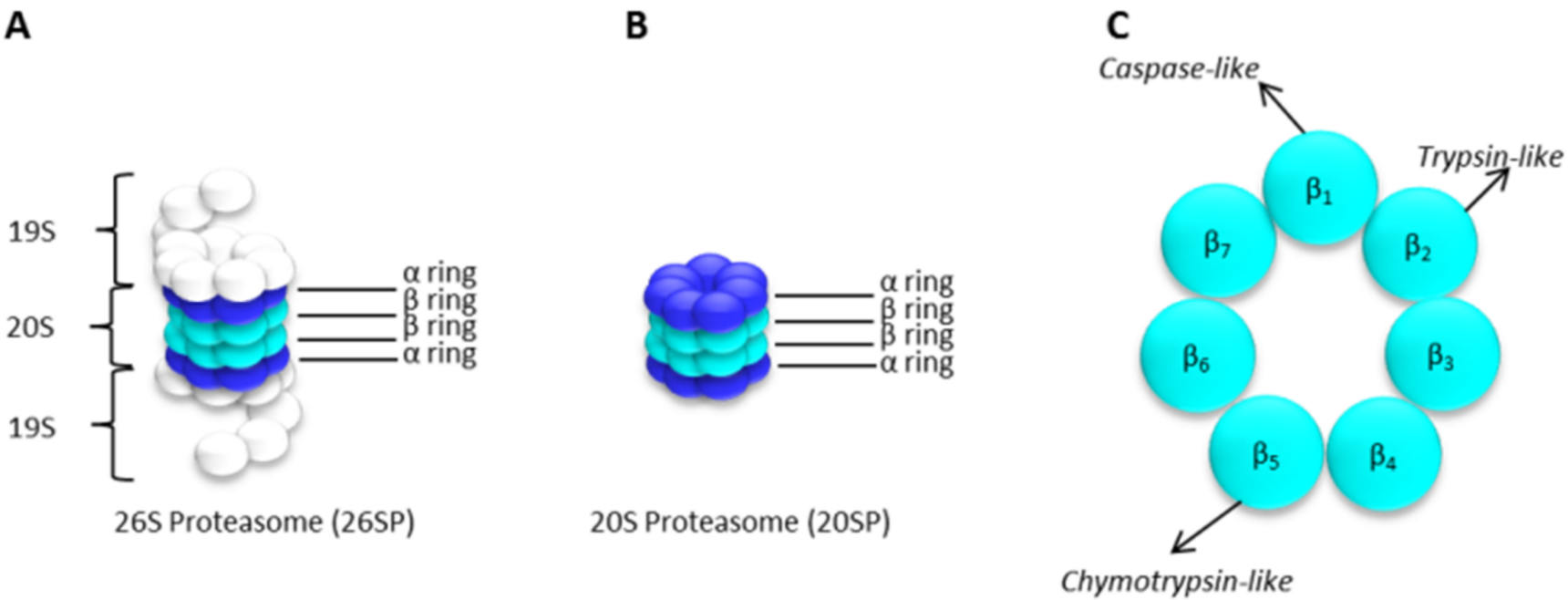
The 20S Proteasome Core Particle
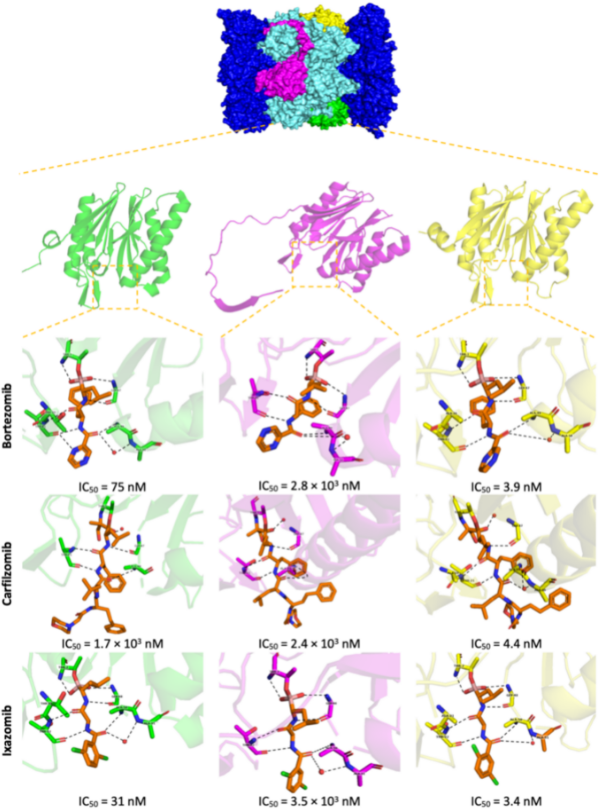
3. Proteasome Inhibitors (PIs) in Cancer Therapy
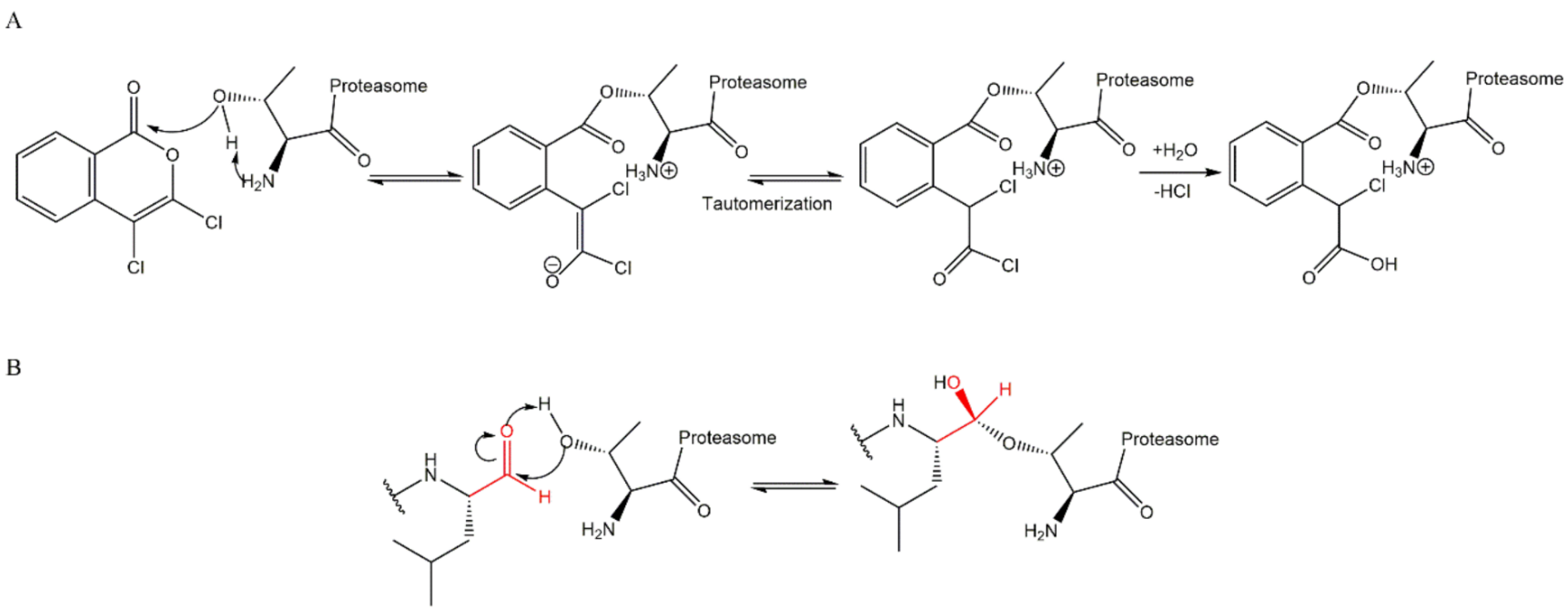
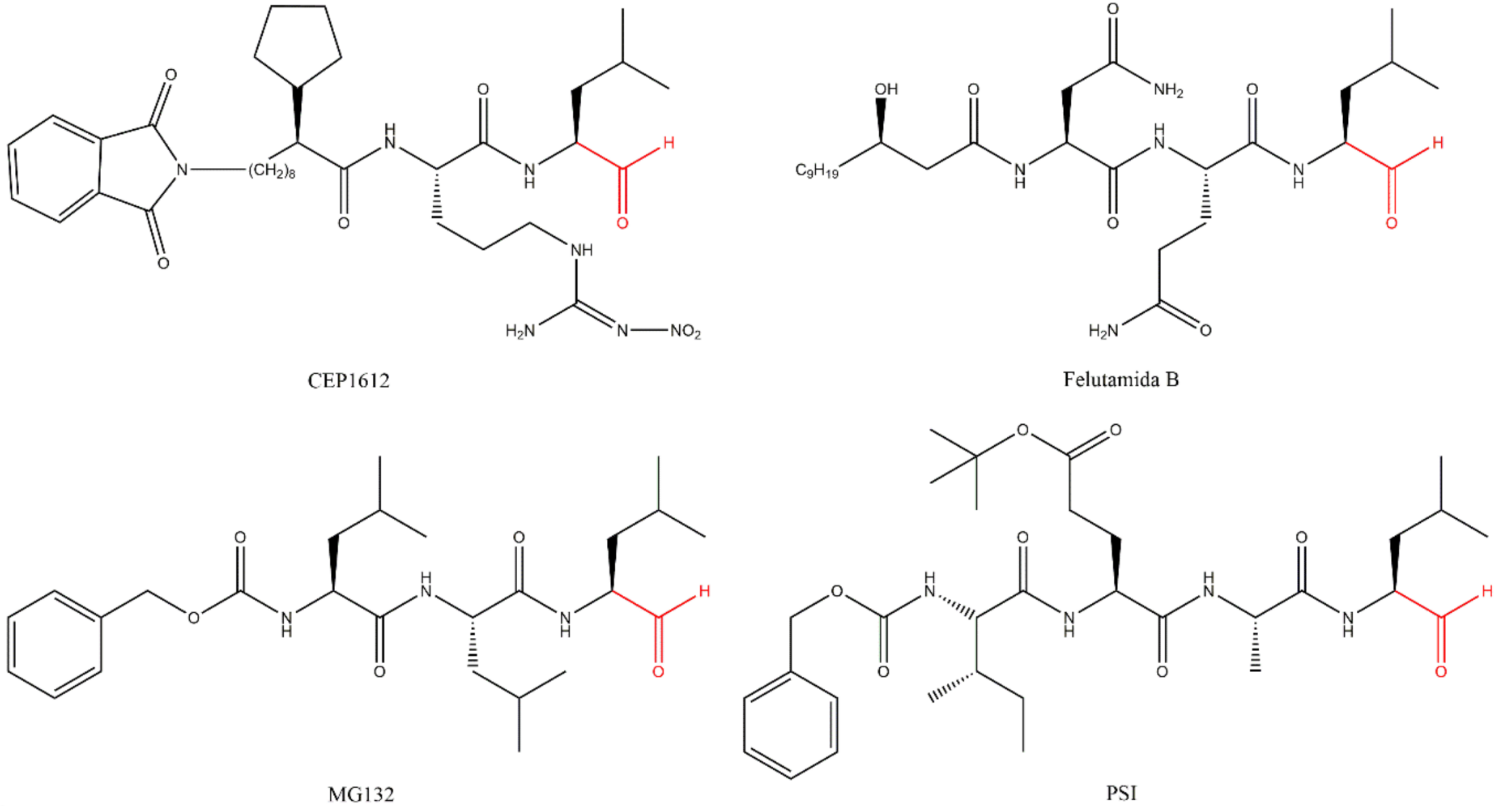
3.1. Aldehydes
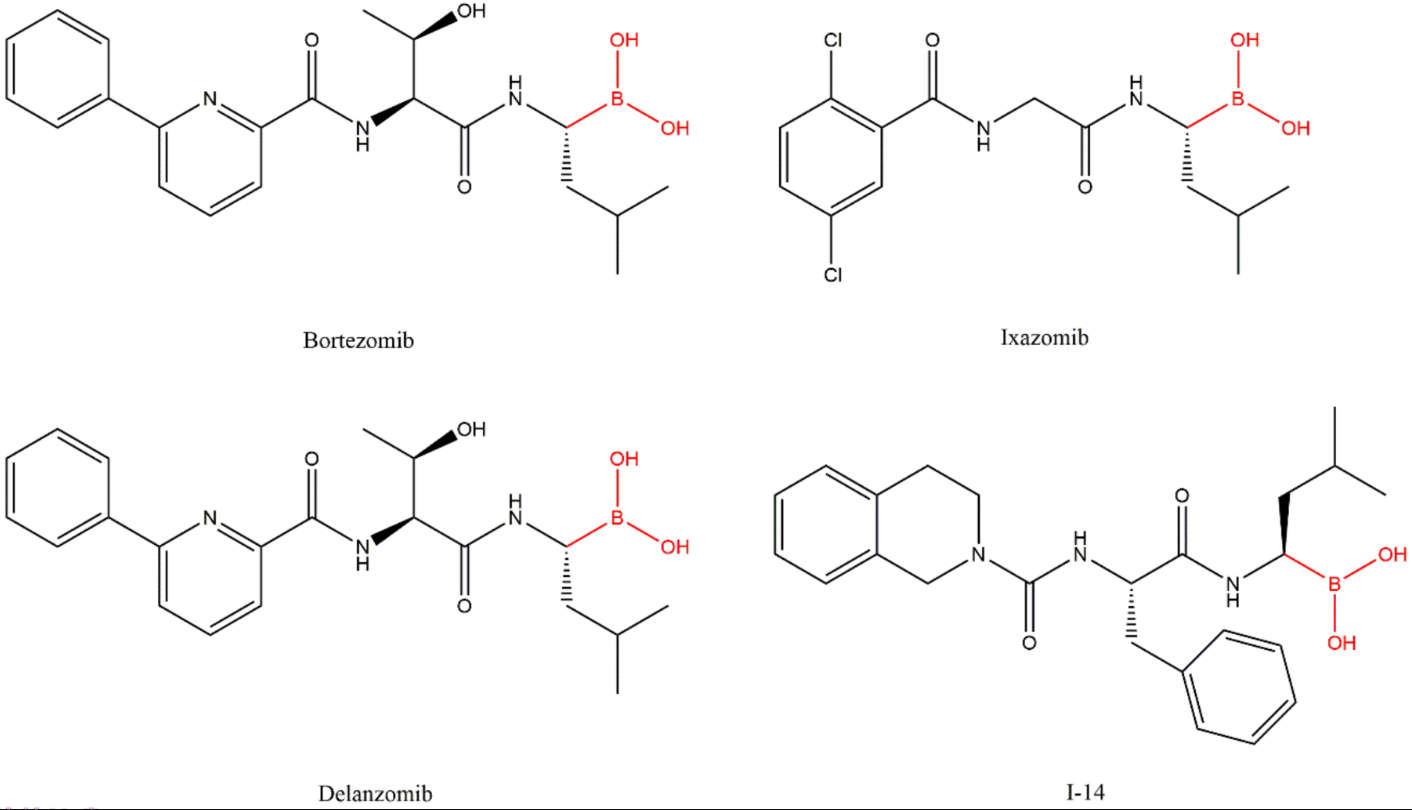
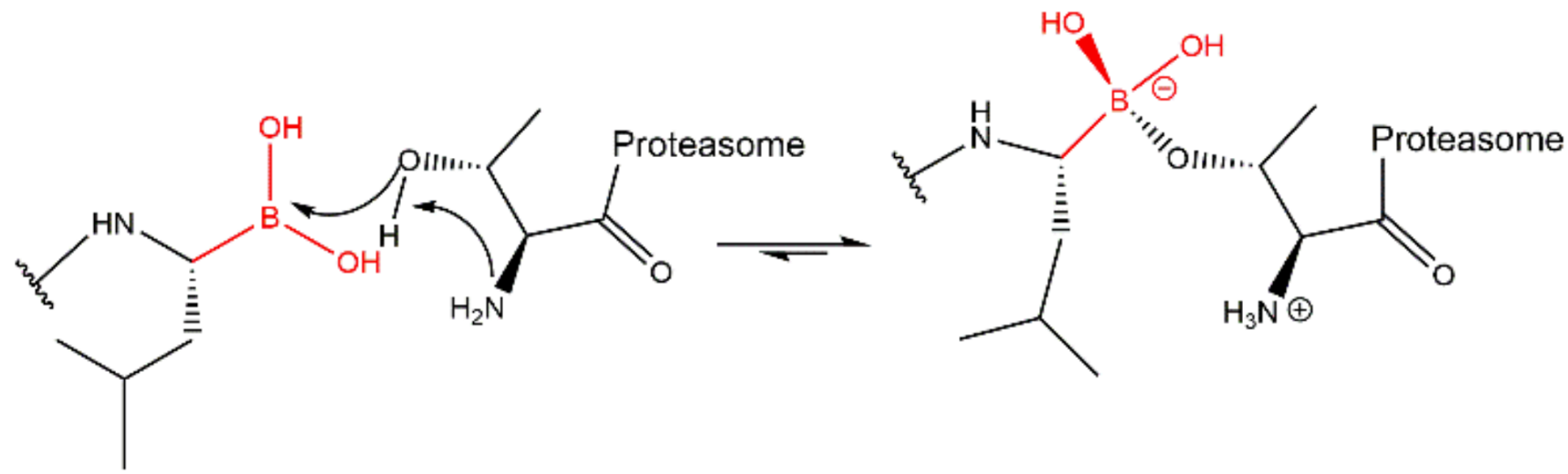
3.2. Boronates
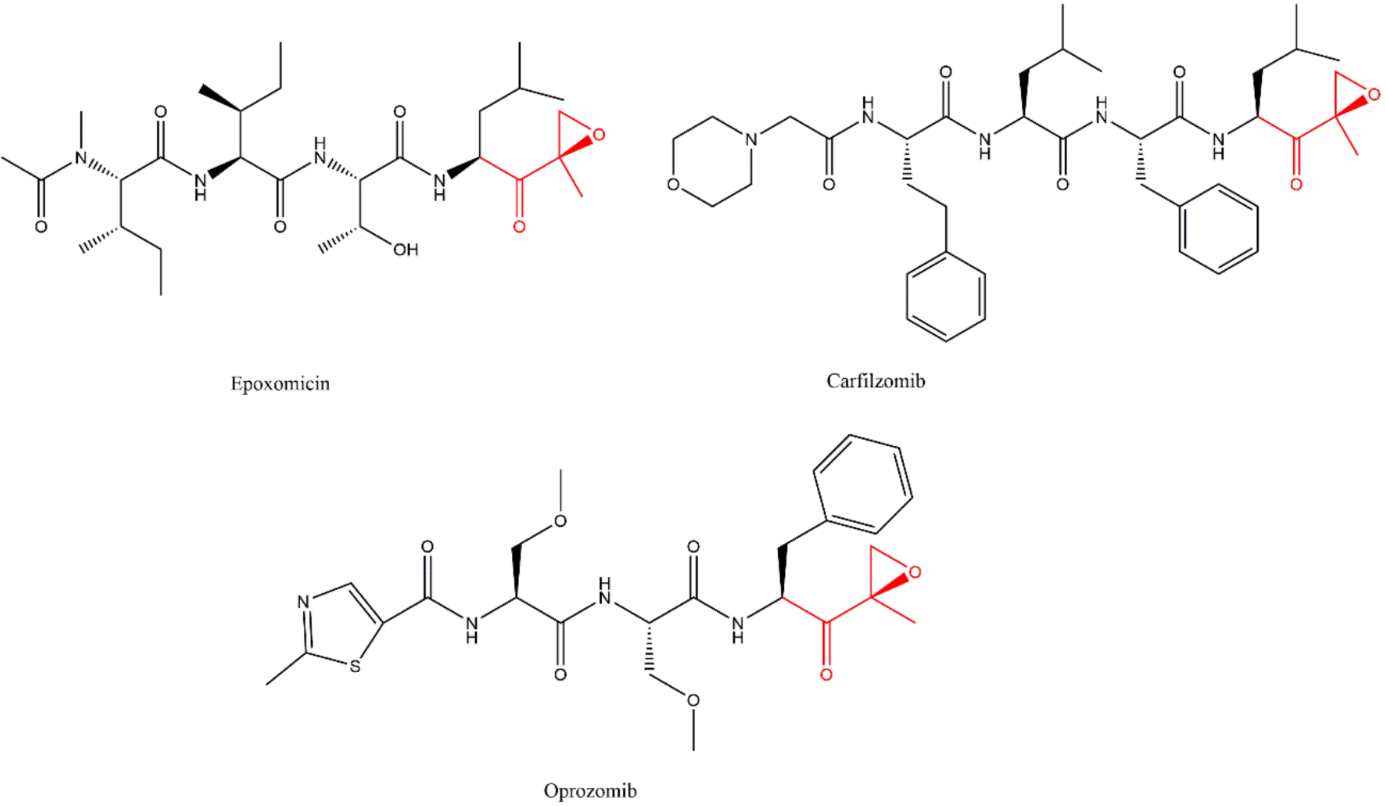
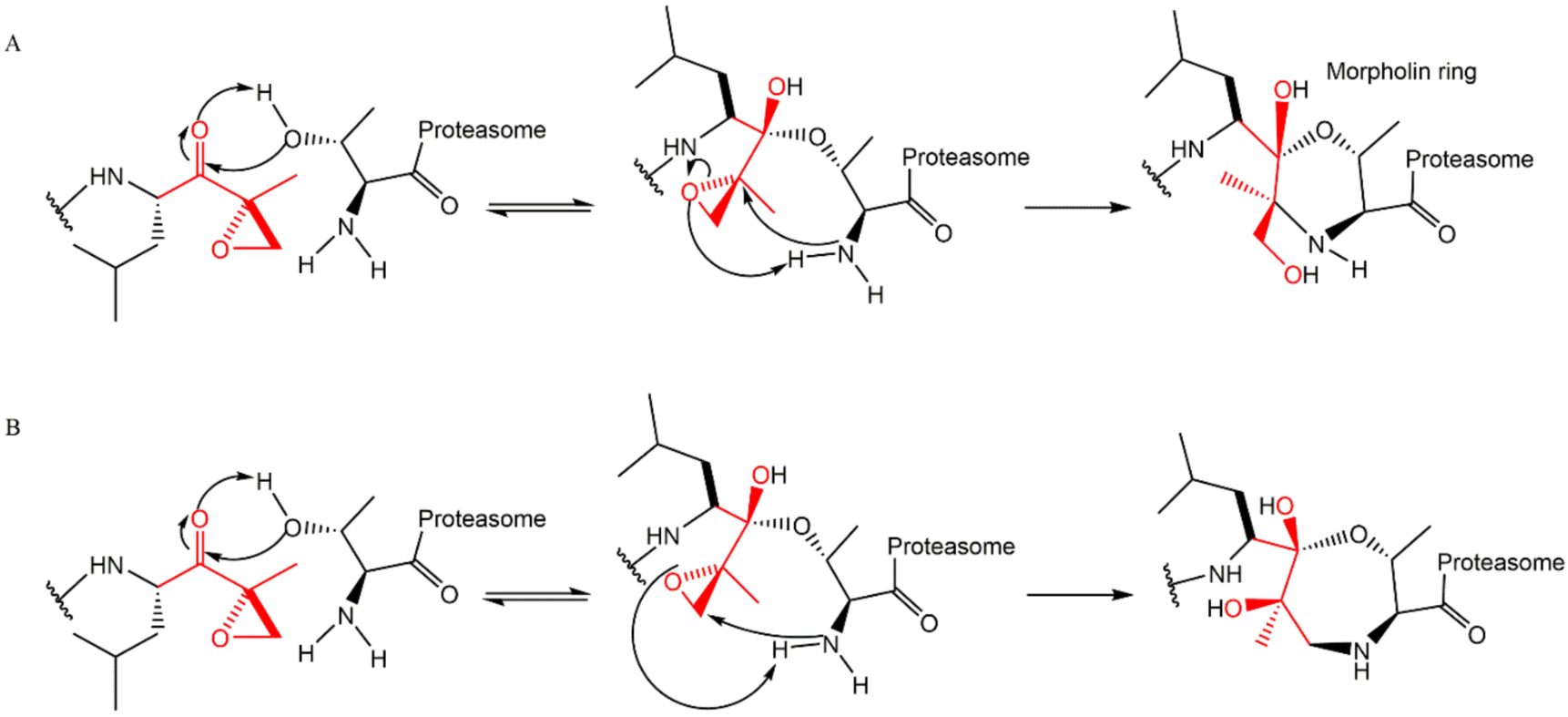
3.3. α’,β’-Epoxyketones
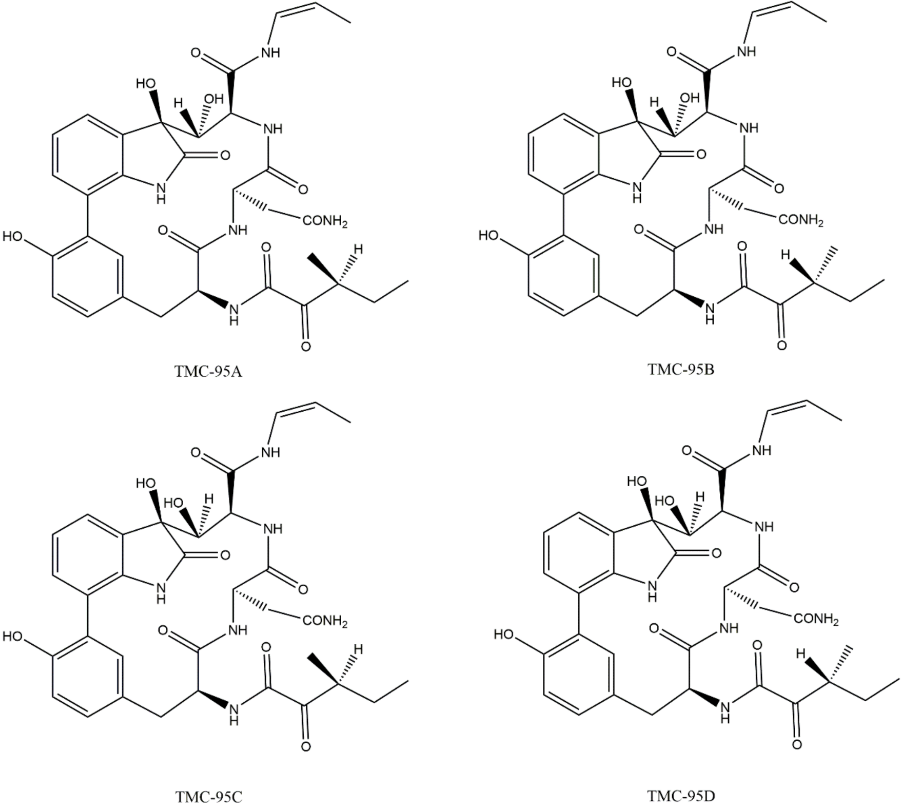
3.4. Non-Covalent Macrocyclics
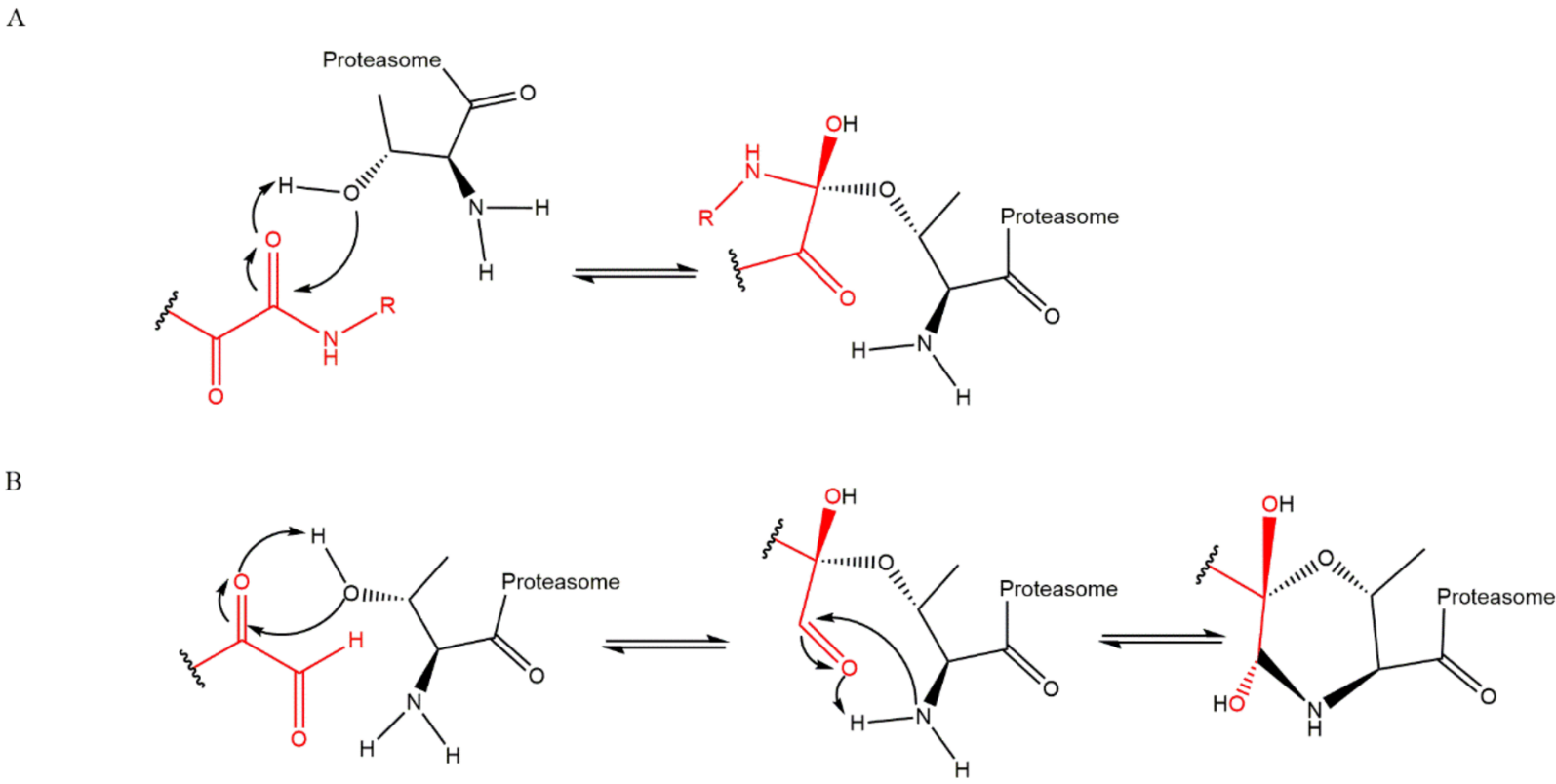
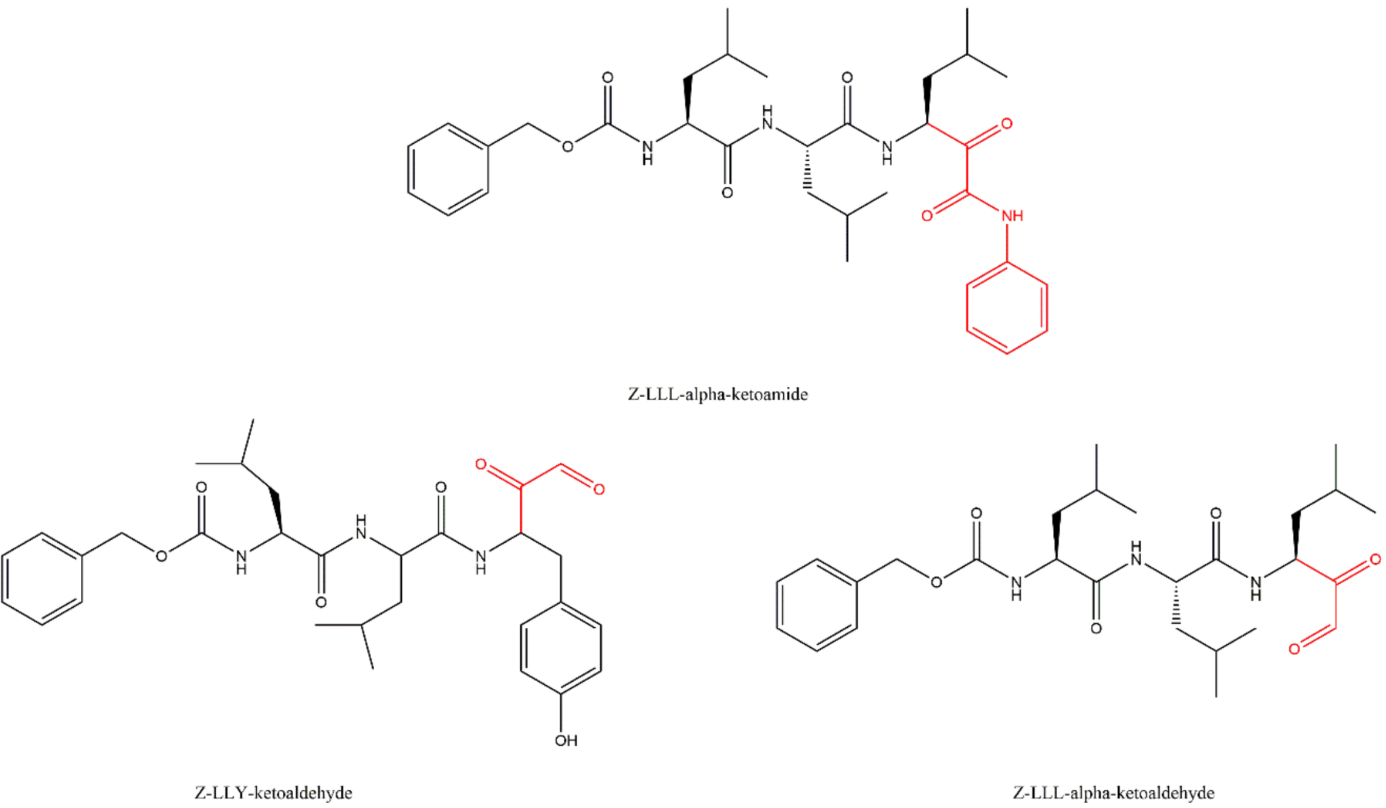
3.5. α-Ketoaldehydes and α-Ketoamides
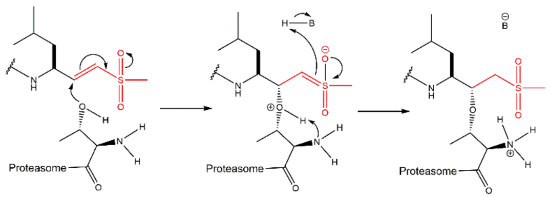
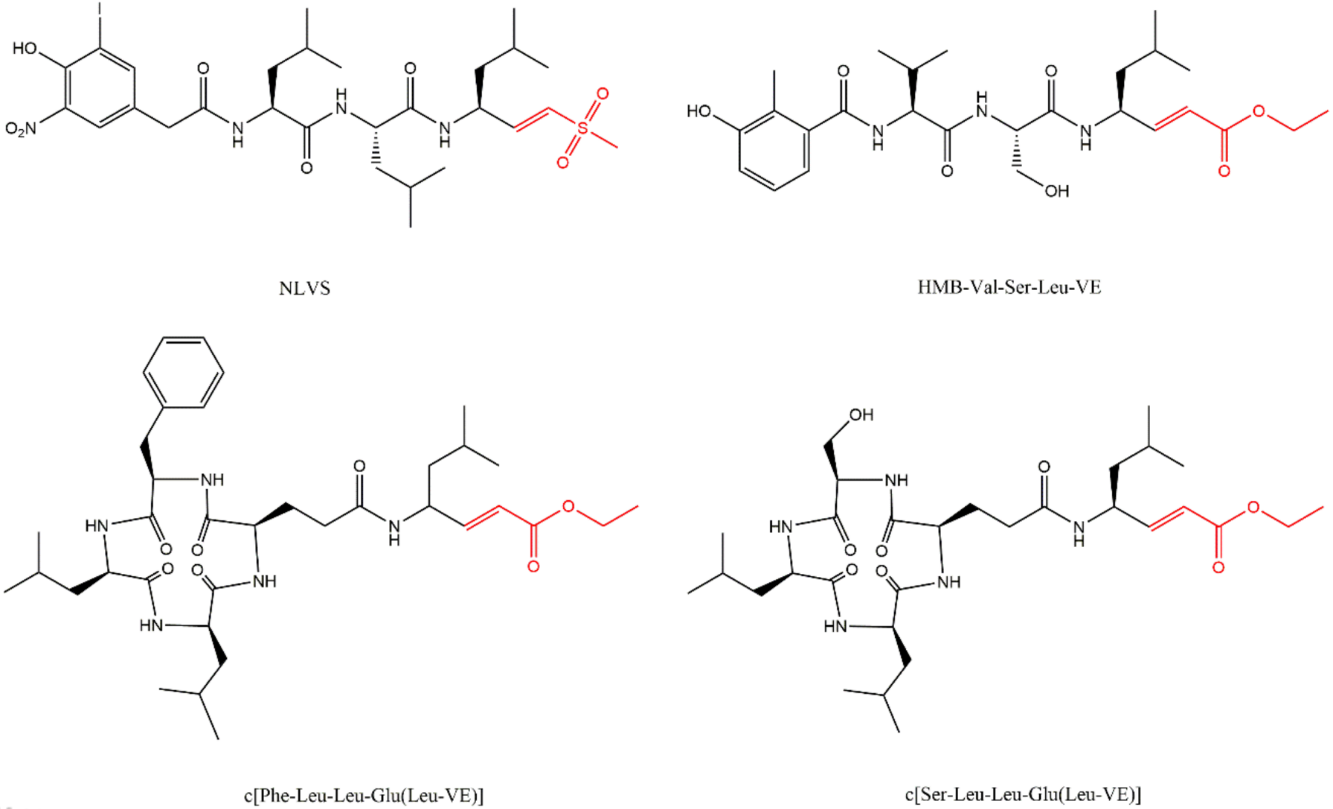
3.6. Peptide Vinyl Derivatives
The vinyl peptide derivatives have electron withdrawing groups (sulfone or ester) in the C-terminal which behave as Michael acceptors of the catalytic Thr1 hydroxyl group, promoting the formation of a covalent irreversible bond (Figure 15) [61].
3.7. β-Lactones
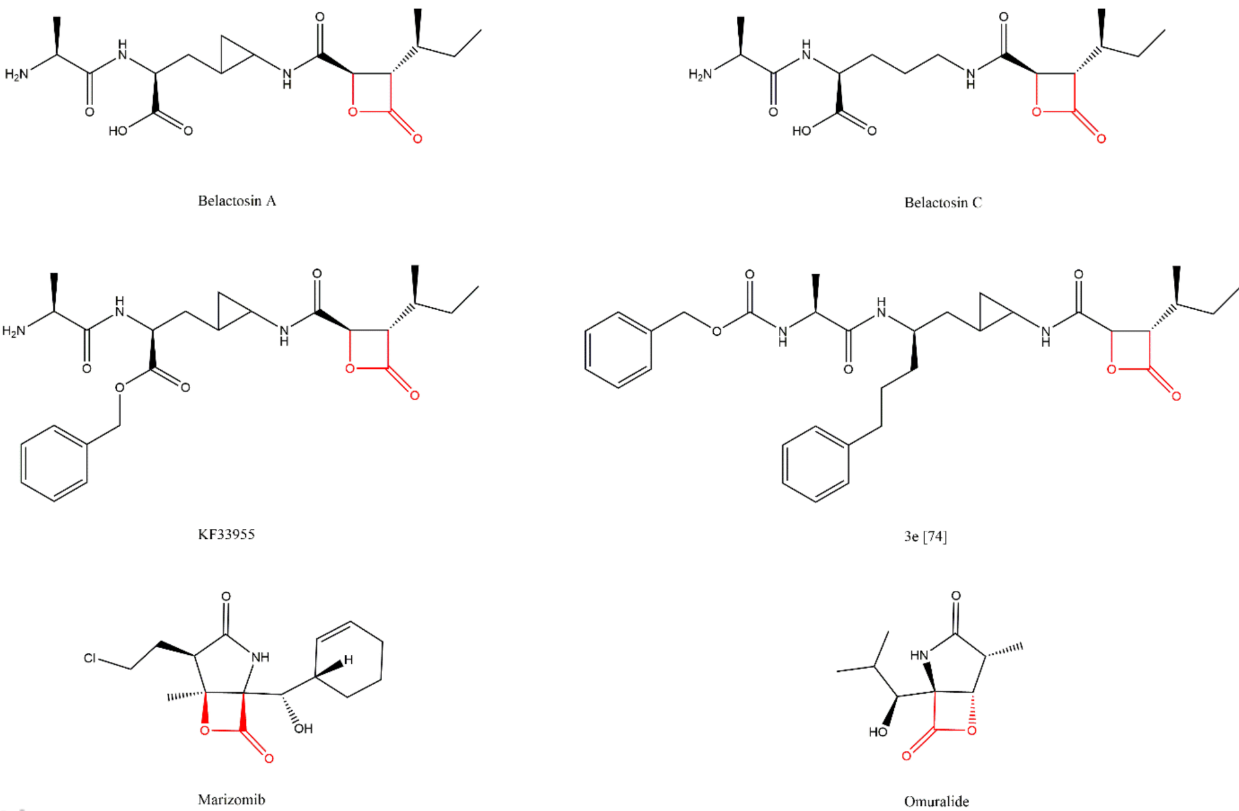
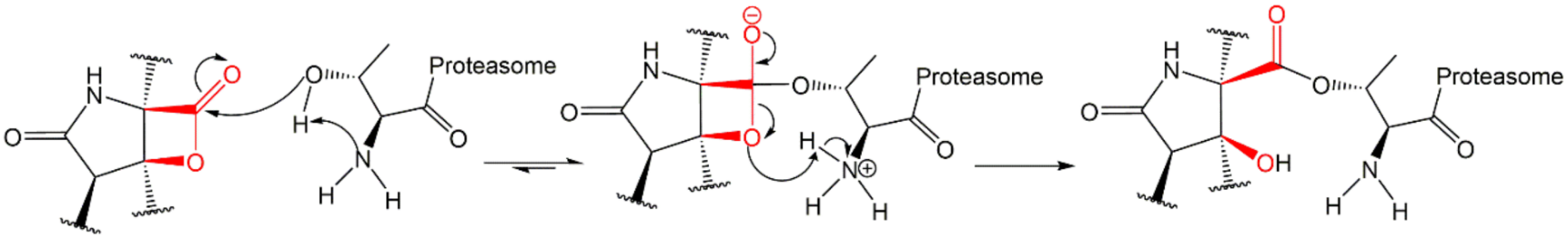
3.8. Syrbactins
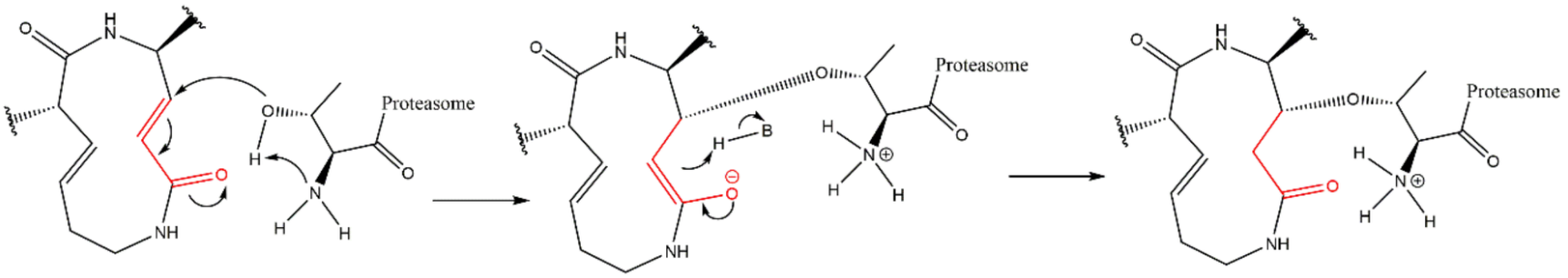
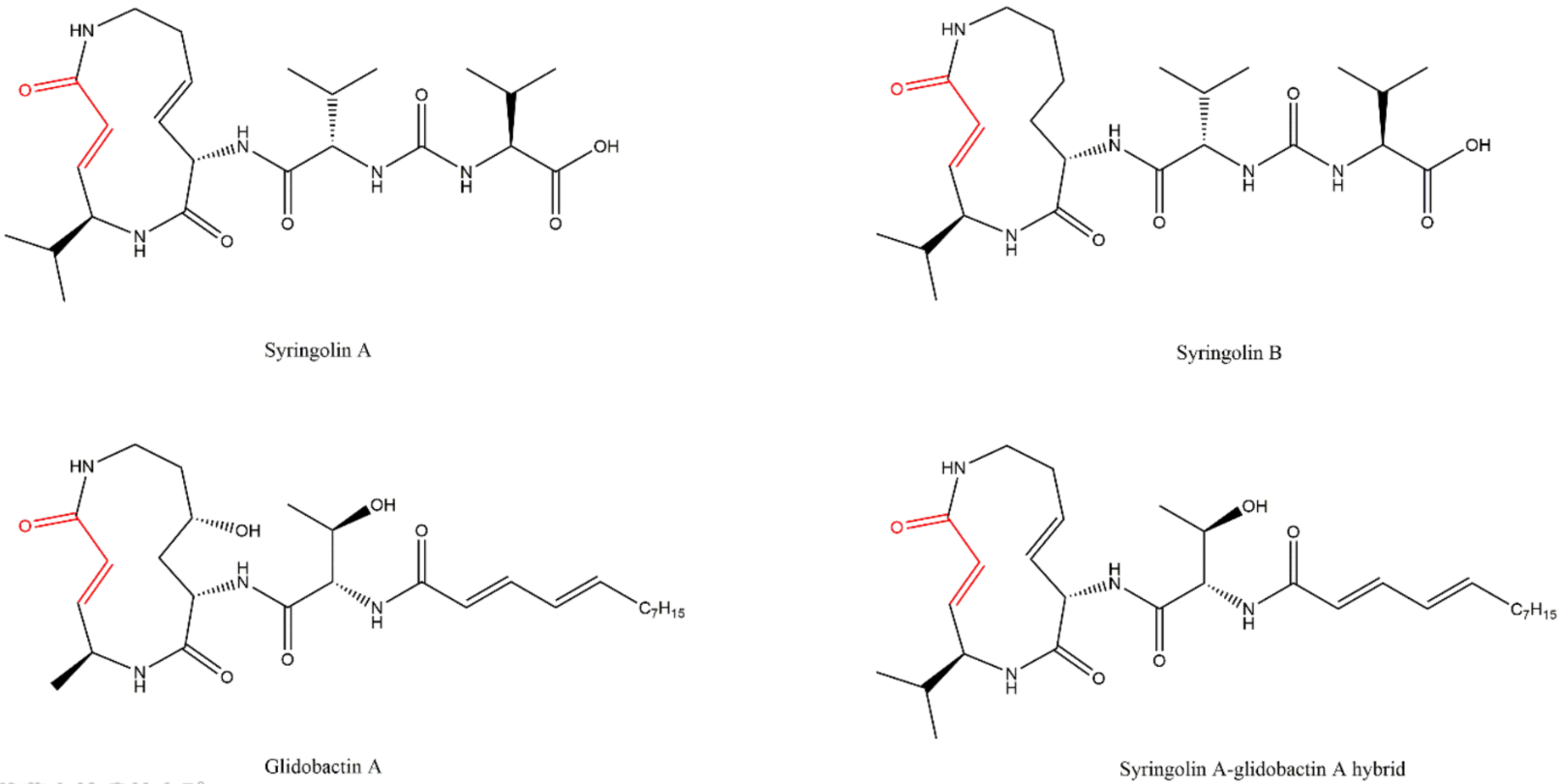
4. Resistance Mechanisms to Proteasome Inhibitors (PIs)
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479.
- Obrist, F.; Manic, G.; Kroemer, G.; Vitale, I.; Galluzzi, L. Trial Watch: Proteasomal inhibitors for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e974463.
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433.
- EMA Ninlaro®: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/ninlaro-epar-product-information_en.pdf (accessed on 5 April 2021).
- EMA Kyprolis®: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/kyprolis-epar-product-information_en.pdf (accessed on 5 April 2021).
- FDA Highlights of Prescribing Information Velcade® (Bortezomib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/206927s000lbl.pdf (accessed on 5 April 2021).
- FDA Highlights of Prescribing Information Ninlaro® (Ixazomib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208462s010lbl.pdf (accessed on 5 April 2021).
- FDA Highlights of Prescribing Information Kyprolis® (Carfilzomib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202714s030lbl.pdf (accessed on 5 April 2021).
- EMA Velcade: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/velcade-epar-product-information_en.pdf (accessed on 5 April 2021).
- Cancer Stat Facts: Myeloma. Available online: https://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 24 February 2022).
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020.
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Investig. 2012, 122, 3456–3463.
- Offidani, M.; Corvatta, L.; Morè, S.; Olivieri, A. Novel experimental drugs for treatment of multiple myeloma. J. Exp. Pharmacol. 2021, 13, 245–264.
- Goldman-Mazur, S.; Kumar, S.K. Current approaches to management of high-risk multiple myeloma. Am. J. Hematol. 2021.
- Roué, G.; Sola, B. Management of drug resistance in mantle cell lymphoma. Cancers 2020, 12, 1565.
- Pérez-Galán, P.; Dreyling, M.; Wiestner, A. Mantle cell lymphoma: Biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 2011, 117, 26–38.
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428.
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases. Neuron 2003, 40, 427–446.
- Thompson, S.; Loftus, L.; Ashley, M.; Meller, R. Ubiquitin–proteasome system as a modulator of cell fate. Curr. Opin. Pharmacol. 2008, 8, 90–95.
- Bhat, K.P.; Greer, S.F. Proteolytic and non-proteolytic roles of ubiquitin and the ubiquitin proteasome system in transcriptional regulation. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 150–155.
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96.
- Kim, H.M.; Yu, Y.; Cheng, Y. Structure characterization of the 26S proteasome. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 67–79.
- Orlowski, N.; Wilk, S. A multicatalytical protease complex from pituitary that forms enkephalin and enkephalin containing peptides. Biochem. Biophys. Res. Commun. 1981, 101, 814–822.
- Young, P.; Deveraux, Q.; Beal, R.E.; Pickart, C.M.; Rechsteiner, M. Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J. Biol. Chem. 1998, 273, 5461–5467.
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407.
- Harer, S.L.; Bhatia, M.S.; Bhatia, N.M. Proteasome inhibitors mechanism; source for design of newer therapeutic agents. J. Antibiot. 2012, 65, 279–288.
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758.
- Borissenko, L.; Groll, M. 20S proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717.
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chemie Int. Ed. 2012, 51, 8708–8720.
- Zwickl, P.; Baumeister, W. (Eds.) The Proteasome—Ubiquitin Protein Degradation Pathway; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2002; Volume 268, ISBN 978-3-642-63971-5.
- Guedes, R.A.; Serra, P.; Salvador, J.; Guedes, R. Computational approaches for the discovery of human proteasome inhibitors: An overview. Molecules 2016, 21, 927.
- Moore, B.S.; Eustáquio, A.S.; McGlinchey, R.P. Advances in and applications of proteasome inhibitors. Curr. Opin. Chem. Biol. 2008, 12, 434–440.
- Adams, J. (Ed.) Proteasome Inhibitors in Cancer Therapy; Humana Press: Totowa, NJ, USA, 2004; ISBN 978-1-61737-452-4.
- Dou, Q.; Zonder, J. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536.
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253.
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 2011, 5, 101–110.
- Achanta, G. A boronic-chalcone derivative exhibits potent anti-cancer activity through inhibition of the proteasome. Mol. Pharmacol. 2006, 579, 3932–3940.
- Han, L.-Q.; Yuan, X.; Wu, X.-Y.; Li, R.-D.; Xu, B.; Cheng, Q.; Liu, Z.-M.; Zhou, T.-Y.; An, H.-Y.; Wang, X.; et al. Urea-containing peptide boronic acids as potent proteasome inhibitors. Eur. J. Med. Chem. 2017, 125, 925–939.
- Ruschak, A.M.; Slassi, M.; Kay, L.E.; Schimmer, A.D. Novel proteasome inhibitors to overcome bortezomib resistance. JNCI J. Natl. Cancer Inst. 2011, 103, 1007–1017.
- ClinicalTrials.gov. Phase I Study of the Proteosome Inhibitor CEP 18770 in Patients with Solid Tumours or Non-Hodgkin’s Lymphomas. Available online: https://www.clinicaltrials.gov/ct2/show/study/NCT00572637?term=delanzomib&rank=3 (accessed on 28 February 2017).
- Gallerani, E.; Zucchetti, M.; Brunelli, D.; Marangon, E.; Noberasco, C.; Hess, D.; Delmonte, A.; Martinelli, G.; Böhm, S.; Driessen, C.; et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer 2013, 49, 290–296.
- ClinicalTrials.gov. Study to Determine the Maximum Tolerated Dose and Evaluate the Efficacy and Safety of CEP-18770 (Delanzomib) in Patients with Relapsed Multiple Myeloma Refractory to the Most Recent Therapy. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01023880?term=delanzomib&rank=1 (accessed on 28 February 2017).
- Vogl, D.T.; Martin, T.G.; Vij, R.; Hari, P.; Mikhael, J.R.; Siegel, D.; Wu, K.L.; Delforge, M.; Gasparetto, C. Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leuk. Lymphoma 2017, 58, 1872–1879.
- ClinicalTrials.gov. Delanzomib (CEP-18770) in Combination with Lenalidomide and Dexamethasone in Relapsed or Refractory Multiple Myeloma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01348919?term=delanzomib&rank=2 (accessed on 28 February 2017).
- ClinicalTrials.gov. A Study of Oprozomib, Melphalan, and Prednisone in Transplant Ineligible Patients with Newly Diagnosed Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02072863?cond=oprozomib&rank=6 (accessed on 5 October 2017).
- ClinicalTrials.gov. Phase 1 Study of Oprozomib Administered Orally in Patients with Advanced Refractory or Recurrent Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01129349?cond=oprozomib&rank=5 (accessed on 5 October 2017).
- ClinicalTrials.gov. Study of Oprozomib and Dexamethasone, in Combination with Lenalidomide or Oral Cyclophosphamide in Patients With Newly Diagnosed Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT01881789?recrs=d&cond=oprozomib&rank=1 (accessed on 5 October 2017).
- ClinicalTrials.gov. Phase 1b/2, Multicenter, Open-label Study of Oprozomib and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT01832727?recrs=d&cond=oprozomib&rank=2 (accessed on 5 October 2017).
- ClinicalTrials.gov. Open-label Study of the Safety and Activity of Oprozomib in Patients with Hematologic Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT01416428?recrs=d&cond=oprozomib&rank=3 (accessed on 5 October 2017).
- ClinicalTrials.gov. A Study of Oprozomib, Pomalidomide, and Dexamethasone in Subjects with Primary Refractory or Relapsed and Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT01999335?recrs=d&cond=oprozomib&rank=4 (accessed on 5 October 2017).
- ClinicalTrials.gov. A Phase 1 Study of Oprozomib to Assess Food Effect, Drug-Drug Interaction with Midazolam, and Safety and Tolerability in Patients with Advanced Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02244112?recrs=d&cond=oprozomib&rank=5 (accessed on 5 October 2017).
- ClinicalTrials.gov. Phase 1b Study Evaluating OPomD in Relapsed or Refractory Multiple Myeloma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02939183?term=Oprozomib&rank=9 (accessed on 28 February 2017).
- Basmadjian, C.; Zhao, Q.; Bentouhami, E.; Djehal, A.; Nebigil, C.G.; Johnson, R.A.; Serova, M.; de Gramont, A.; Faivre, S.; Raymond, E.; et al. Cancer wars: Natural products strike back. Front. Chem. 2014, 2, 20.
- Groll, M.; Nazif, T.; Huber, R.; Bogyo, M. Probing structural determinants distal to the site of hydrolysis that control substrate specificity of the 20S proteasome. Chem. Biol. 2002, 9, 655–662.
- Gräwert, M.A.; Gallastegui, N.; Stein, M.; Schmidt, B.; Kloetzel, P.-M.; Huber, R.; Groll, M. Elucidation of the α-keto-aldehyde binding mechanism: A lead structure motif for proteasome inhibition. Angew. Chemie Int. Ed. 2011, 50, 542–544.
- Stein, M.L.; Cui, H.; Beck, P.; Dubiella, C.; Voss, C.; Krüger, A.; Schmidt, B.; Groll, M. Systematic comparison of peptidic proteasome inhibitors highlights the α-ketoamide electrophile as an auspicious reversible lead motif. Angew. Chemie Int. Ed. 2014, 53, 1679–1683.
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20 S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598.
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora Montagnei Sacc. TC 1093. taxonomy, production, isolation, and biological activities. J. Antibiot. 2000, 53, 105–109.
- Kohno, J.; Koguchi, Y.; Nishio, M.; Nakao, K.; Kuroda, M.; Shimizu, R.; Ohnuki, T.; Komatsubara, S. Structures of TMC-95A-D: Novel proteasome inhibitors from Apiospora montagnei Sacc. TC 1093. J. Org. Chem. 2000, 65, 990–995.
- Groll, M.; Koguchi, Y.; Huber, R.; Kohno, J. Crystal structure of the 20 S proteasome:TMC-95A complex: A non-covalent proteasome inhibitor. J. Mol. Biol. 2001, 311, 543–548.
- Génin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: Recent advances and new perspectives in medicinal chemistry. Curr. Top. Med. Chem. 2010, 10, 232–256.
- Nazif, T.; Bogyo, M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 2967–2972.
- Masters, E.I. Structural Studies of the Proteasome and Implications for Proteasome Activation. Ph.D. Thesis, Department of Biochemistry, University of Utah, Salt Lake City, UT, USA, 2008.
- Marastoni, M.; Baldisserotto, A.; Cellini, S.; Gavioli, R.; Tomatis, R. Peptidyl vinyl ester derivatives: New class of selective inhibitors of proteasome trypsin-like activity. J. Med. Chem. 2005, 48, 5038–5042.
- Marastoni, M.; Baldisserotto, A.; Trapella, C.; Gavioli, R.; Tomatis, R. Synthesis and biological evaluation of new vinyl ester pseudotripeptide proteasome inhibitors. Eur. J. Med. Chem. 2006, 41, 978–984.
- Marastoni, M.; Baldisserotto, A.; Canella, A.; Gavioli, R.; De Risi, C.; Pollini, G.P.; Tomatis, R. Arecoline tripeptide inhibitors of proteasome. J. Med. Chem. 2004, 47, 1587–1590.
- Baldisserotto, A.; Destro, F.; Vertuani, G.; Marastoni, M.; Gavioli, R.; Tomatis, R. N-Terminal-prolonged vinyl ester-based peptides as selective proteasome β1 subunit inhibitors. Bioorg. Med. Chem. 2009, 17, 5535–5540.
- Baldisserotto, A.; Marastoni, M.; Fiorini, S.; Pretto, L.; Ferretti, V.; Gavioli, R.; Tomatis, R. Vinyl ester-based cyclic peptide proteasome inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1849–1854.
- Baldisserotto, A.; Marastoni, M.; Gavioli, R.; Tomatis, R. New cyclic peptide proteasome inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1966–1969.
- Franceschini, C.; Trapella, C.; Sforza, F.; Gavioli, R.; Marastoni, M. Synthesis and biological properties of C-terminal vinyl ketone pseudotripeptides. J. Enzyme Inhib. Med. Chem. 2013, 28, 560–564.
- Baldisserotto, A.; Ferretti, V.; Destro, F.; Franceschini, C.; Marastoni, M.; Gavioli, R.; Tomatis, R. α,β-unsaturated N -acylpyrrole peptidyl derivatives: New proteasome inhibitors. J. Med. Chem. 2010, 53, 6511–6515.
- Asai, A.; Hasegawa, A.; Ochiai, K.; Yamashita, Y.; Mizukami, T. Belactosin A, a Novel antitumor antibiotic acting on cyclin/CDK mediated cell cycle regulation, produced by Streptomyces sp. J. Antibiot. 2000, 53, 81–83.
- Asai, A.; Tsujita, T.; Sharma, S.V.; Yamashita, Y.; Akinaga, S.; Funakoshi, M.; Kobayashi, H.; Mizukami, T. A new structural class of proteasome inhibitors identified by microbial screening using yeast-based assay. Biochem. Pharmacol. 2004, 67, 227–234.
- Kawamura, S.; Unno, Y.; List, A.; Mizuno, A.; Tanaka, M.; Sasaki, T.; Arisawa, M.; Asai, A.; Groll, M.; Shuto, S. Potent proteasome inhibitors derived from the unnatural cis-cyclopropane isomer of Belactosin A: Synthesis, biological activity, and mode of action. J. Med. Chem. 2013, 56, 3689–3700.
- Nakamura, H.; Watanabe, M.; Ban, H.S.; Nabeyama, W.; Asai, A. Synthesis and biological evaluation of boron peptide analogues of Belactosin C as proteasome inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3220–3224.
- COMP Marizomib for the Treatment of Plasma Cell Myeloma. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/09/WC500171952.pdf (accessed on 6 April 2017).
- COMP Marizomib for the Treatment of Glioma. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/2119-public-summary-opinion-orphan-designation-marizomib-treatment-glioma_en.pdf (accessed on 21 February 2020).
- Triphase Accelerator Corporation Marizomib. Available online: http://triphaseco.com/marizomib/ (accessed on 2 October 2017).
- Triphase Accelerator Corporation Triphase Receives FDA Orphan Drug Designation for Marizomib in Multiple Myeloma. Available online: http://triphaseco.com/wp-content/uploads/2014/02/20140226_Triphase_orphan_drug_news-release_final.pdf (accessed on 4 October 2018).
- Triphase Accelerator Corporation Triphase Accelerator Corporation Announces FDA Orphan Drug Designation for Marizomib in Malignant Glioma. Available online: http://triphaseco.com/wp-content/uploads/2015/11/MRZ-Orphan-Press-Release-11192015_FINAL.pdf (accessed on 4 October 2018).
- ClinicalTrials.gov. Phase 1 Trial of Marizomib Alone and in Combination with Panobinostat for Children with Diffuse Intrinsic Pontine Glioma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04341311?cond=marizomib&draw=3&rank=4 (accessed on 5 April 2021).
- ClinicalTrials.gov. Phase 2 Clinical Trial of NPI-0052 in Patients with Relapsed or Relapsed/Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT00461045?term=marizomib&draw=2&rank=7 (accessed on 21 February 2020).
- ClinicalTrials.gov. ABI-009 (Nab-Rapamycin) in Recurrent High Grade Glioma and Newly Diagnosed Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03463265?term=marizomib&draw=2&rank=10 (accessed on 20 February 2020).
- ClinicalTrials.gov. Phase 1 Clinical Trial of NPI-0052 in Patients with Advanced Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT00629473?cond=marizomib&rank=5 (accessed on 6 October 2017).
- ClinicalTrials.gov. NPI-0052 and Vorinostat in Patients with Non-small Cell Lung Cancer, Pancreatic Cancer, Melanoma or Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00667082?term=marizomib&draw=2&rank=3 (accessed on 21 February 2020).
- ClinicalTrials.gov. Combination Study of Pomalidomide, Marizomib, and Low-Dose Dexamethasone in Relapsed and Refractory Multiple Myeloma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02103335?term=Marizomib&rank=3 (accessed on 28 February 2017).
- ClinicalTrials.gov. Stage 1: Marizomib + Bevacizumab in WHO Gr IV GBM; Stage 2: Marizomib Alone; Stage 3: Combination of Marizomib and Bevacizumab. Available online: https://clinicaltrials.gov/ct2/show/NCT02330562?cond=marizomib&rank=3 (accessed on 6 October 2017).
- ClinicalTrials.gov. Study of Marizomib With Temozolomide and Radiotherapy in Patients with Newly Diagnosed Brain Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02903069?cond=marizomib&rank=1 (accessed on 6 October 2017).
- ClinicalTrials.gov. A Phase III Trial of With Marizomib in Patients with Newly Diagnosed Glioblastoma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03345095?term=NPI-0052&draw=2&rank=9 (accessed on 5 April 2021).
- ClinicalTrials.gov. Marizomib for Recurrent Low-Grade and Anaplastic Supratentorial, Infratentorial, and Spinal Cord Ependymoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03727841?term=marizomib&draw=2&rank=2 (accessed on 20 February 2020).
- ClinicalTrials.gov. Phase 1 Clinical Trial of NPI-0052 in Patients with Advanced Solid Tumor Malignancies or Refractory Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00396864?cond=marizomib&rank=7 (accessed on 6 October 2017).
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase I clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma: Study NPI-0052-102 final results. Clin. Cancer Res. 2016, 22, 4559–4566.
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 2012, 30, 2303–2317.
- Spencer, A.; Laubach, J.P.; Zonder, J.A.; Badros, A.Z.; Harrison, S.; Khot, A.; Chauhan, D.; Anderson, K.C.; Reich, S.D.; Trikha, M.; et al. Phase 1, multicenter, open-label, combination study (NPI-0052-107; NCT02103335) of pomalidomide (POM), marizomib (MRZ, NPI-0052), and low-dose dexamethasone (LD-DEX) in patients with relapsed and refractory multiple myeloma. Blood 2015, 126, 4220.
- Spencer, A.; Harrison, S.; Laubach, J.P.; Zonder, J.; Badros, A.Z.; Bergin, K.; Khot, A.; Zimmerman, T.; Anderson, K.C.; MacLaren, A.; et al. Pmd-107: Marizomib, pomalidomide and low dose-dexamethasone combination study in relapsed/refractory multiple myeloma (NCT02103335): Full enrollment results from a phase-1 multicenter, open label study. Blood 2016, 128, 3326.
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib alone or in combination with bevacizumab in patients with recurrent glioblastoma: Phase I/II clinical trial data. Neuro-Oncol. Adv. 2021, 3, 1–10.
- Mason, W.P.; Kesari, S.; Stupp, R.; Aregawi, D.G.; Piccioni, D.E.; Roth, P.; Desjardins, A.; Reich, S.D.; Casadebaig, M.-L.; Elias, I.; et al. Full enrollment results from an extended phase I, multicenter, open label study of marizomib (MRZ) with temozolomide (TMZ) and radiotherapy (RT) in newly diagnosed glioblastoma (GBM). J. Clin. Oncol. 2019, 37, 2021.
- ClinicalTrials.gov. Marizomib Central Nervous System (CNS). Available online: https://clinicaltrials.gov/ct2/show/NCT05050305?term=marizomib&draw=1&rank=4 (accessed on 22 February 2022).
- Clerc, J.; Groll, M.; Illich, D.J.; Bachmann, A.S.; Huber, R.; Schellenberg, B.; Dudler, R.; Kaiser, M. Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 6507–6512.
- Clerc, J.; Schellenberg, B.; Groll, M.; Bachmann, A.S.; Huber, R.; Dudler, R.; Kaiser, M. Convergent synthesis and biological evaluation of syringolin A and derivatives as eukaryotic 20S proteasome inhibitors. Eur. J. Org. Chem. 2010, 2010, 3991–4003.
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115.
- Krahn, D.; Ottmann, C.; Kaiser, M. The chemistry and biology of syringolins, glidobactins and cepafungins (syrbactins). Nat. Prod. Rep. 2011, 28, 1854.
- Archer, C.R.; Groll, M.; Stein, M.L.; Schellenberg, B.; Clerc, J.; Kaiser, M.; Kondratyuk, T.P.; Pezzuto, J.M.; Dudler, R.; Bachmann, A.S. Activity enhancement of the synthetic syrbactin proteasome inhibitor hybrid and biological evaluation in tumor cells. Biochemistry 2012, 51, 6880–6888.
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Bortezomib: Proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 2006, 57, 33–47.
- Orlowski, R.Z.; Kuhn, D.J. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin. Cancer Res. 2008, 14, 1649–1657.
- Dou, Q.P. (Ed.) Resistance to Proteasome Inhibitors in Cancer; Resistance to Targeted Anti-Cancer Therapeutics; Springer International Publishing: Los Angeles, CA, USA, 2014; ISBN 978-3-319-06751-3.
- Suzuki, E.; Demo, S.; Deu, E.; Keats, J.; Arastu-Kapur, S.; Bergsagel, P.L.; Bennett, M.K.; Kirk, C.J. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS ONE 2011, 6, e27996.
- Verbrugge, S.E.; Al, M.; Assaraf, Y.G.; Niewerth, D.; Van Meerloo, J.; Cloos, J.; Van Der Veer, M.; Scheffer, G.L.; Peters, G.J.; Chan, E.T.; et al. Overcoming bortezomib resistance in human B cells by anti-CD20/rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Exp. Hematol. Oncol. 2013, 2, 1.
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin–proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105.
- Vij, R.; Siegel, D.S.; Jagannath, S.; Jakubowiak, A.J.; Stewart, A.K.; McDonagh, K.; Bahlis, N.; Belch, A.; Kunkel, L.A.; Wear, S.; et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br. J. Haematol. 2012, 158, 739–748.
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825.
- Berenson, J.R.; Hilger, J.D.; Yellin, O.; Dichmann, R.; Patel-Donnelly, D.; Boccia, R.V.; Bessudo, A.; Stampleman, L.; Gravenor, D.; Eshaghian, S.; et al. Replacement of bortezomib with carfilzomib for multiple myeloma patients progressing from bortezomib combination therapy. Leukemia 2014, 28, 1529–1536.
- ClinicalTrials.gov. Ixazomib as a Replacement for Carfilzomib and Bortezomib for Multiple Myeloma Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02206425?term=bortezomib&draw=4&rank=669 (accessed on 7 October 2018).
- Berenson, J.R.; Cohen, A.; Spektor, T.M.; Bitran, J.D.; Chen, G.Q.; Moezi, M.M.; Bessudo, A.; Ye, J.Z.; Hager, S.J.; Moss, R.A.; et al. Replacement of ixazomib for relapsed/refractory multiple myeloma patients refractory to a bortezomib or carfilzomib-containing combination therapy. J. Clin. Oncol. 2017, 35, 8013.
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720.
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit 5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499.
- Chauhan, D.; Li, G.; Shringarpure, R.; Podar, K.; Ohtake, Y.; Hideshima, T.; Anderson, K.C. Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res. 2003, 63, 6174–6177.
- Kuhn, D.J.; Berkova, Z.; Jones, R.J.; Woessner, R.; Bjorklund, C.C.; Ma, W.; Davis, R.E.; Lin, P.; Wang, H.; Madden, T.L.; et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012, 120, 3260–3270.
- Cohen, Y.C.; Zada, M.; Wang, S.-Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503.



