 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raffaella Faraonio | -- | 6714 | 2022-04-14 17:58:43 | | | |
| 2 | Catherine Yang | -3378 word(s) | 3336 | 2022-04-15 03:55:35 | | |
Video Upload Options
Cell senescence is critical in diverse aspects of organism life. It represents a physiological program to arrest proliferation of damaged/harmful cells and is now considered an important contributor in aging and age-related pathologies. Non-coding RNAs are endogenous transcripts that govern gene regulatory networks impacting both physiological and pathological events. Among them, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and more recently circular RNAs (circRNAs) are considered crucial regulators of almost all cellular processes, including responses evoked by oxidative stress, such as senescence.
1. Introduction
Forty years ago, Dr. Leonard Hayflick provided the first evidence that primary human cells had a limited replicative capacity in culture (often referred to as the “Hayflick Limit”) [1]. The major determinant of this phenomenon, termed replicative senescence, is the length of telomeres [2]. Cells sense short telomeres as unresolved DNA damage and develop a double-strand DNA break response that irreversibly arrests proliferation [3]. However, telomere shortening is not the only cause of cell senescence. In fact, senescence is rapidly elicited in response to different types of stressors, such as increased oxidative stress [4][5], mitochondrial dysfunctions [6], aberrant oncogene activation [7], ionizing/ultraviolet radiations [8], epigenetic and chromatin perturbations [9] and chemotherapeutic drugs [10]. Of note, most of the senescence-inducing stimuli have been associated to reactive oxygen species (ROS) and Reactive nitrogen species (RNS) increase [11][12].
There are multiple levels of regulation in the signaling network(s) initiating the cell senescence program and sustaining it once activated. Most of these are based on transcriptional and posttranscriptional, as well as epigenetic changes highlighting not only proteins, but also different types of RNAs, including non-coding RNAs (ncRNAs), as crucial components of cell senescence program [13][14][15]. Non-coding RNAs constitute the majority of endogenous transcripts in the cells and among them, the “regulatory” ncRNAs group is linked to gene expression regulation [15][16]. In particular, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and more recently circular RNAs (circRNAs) have been shown to play important roles in the physiopathology of cell and organism senescence [17][18][19][20].
2. Cell Senescence: A Focus on ROS/RNS-Mediated Pathways
The “oxidative stress theory of aging” postulates that cumulative oxidative damages to biomolecules, caused by “a disturbance in the prooxidant–antioxidant balance in favor of the former”, are responsible for the natural ageing process and that this can foster age-related pathological processes [21]. This theory introduced the concept of cell response to redox imbalance and recognized a primary role of ROS/RNS in physio-pathological decline.
2.1. ROS/RNS, Redox Homeostasis and Cell Senescence
ROS and RNS are highly reactive molecules that can originate as byproducts of the aerobic metabolism inside the mitochondrial electron transport chain (ETC) or from dedicated enzymatic reactions, as well as directly through the Fenton reactions [22][23][24][25] (Figure 1, upper panel). H2O2 has a low reactivity and is the main component of signaling cascades leading to changes in protein activities [22][26]. H2O2 is also regarded as the main source of HO•, which is the most reactive ROS provoking non-enzymatic oxidation in all the biomolecules [22].
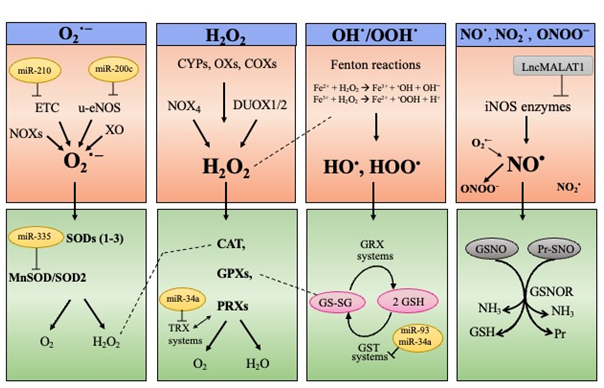
Figure 1. ROS and RNS biological sources (upper panel) and major scavenging pathways (lower panel). ROS comprise the superoxide anion (O2•−), the hydroxyl (HO•), and hydroperoxyl (HOO•) radicals and the non-radical hydrogen peroxide (H2O2). Prevalent RNS are the nitric oxide (NO•), the radical nitrogen dioxide (NO2•), and the peroxynitrite anion (ONOO−). O2•− can originate inside the mitochondrial electron transport chain (ETC) or from enzymatic reactions catalyzed by NAD(P)H oxidases (NOXs), xanthine oxidase (XO) or uncoupling nitric oxide synthase (u-eNOS). H2O2 is produced by NOX4, dual oxidase 1 and 2 (DUOX1/2), cytochrome P450s (CYPs), various oxidases (XOs), cyclooxygenases (COXs), as well as transiently by superoxide dismutase (SOD1–3) isoforms. HO• and HOO• are directly generated through the Fenton reactions. NO• is produced by the enzymes nitric oxide synthases (NOSs), while the peroxynitrite anion (ONOO−) originates from the radical nitrogen dioxide (NO2•) combined with O2•−. miRNAs and long non-coding RNAs affecting expression of ROS/RNS producing enzymes or antioxidant enzymes are reported.
To cope with ROS/RNS toxicity, living cells use sophisticated endogenous antioxidant systems: enzymatic and non-enzymatic scavengers, able to prevent (or quench) oxidative reactions, and consequently, cell physiology degeneration [22][27]. There is an enzymatic network to intercept and scavenge ROS/RNS; examples include: SOD1–3 isoenzymes, catalase, glutathione peroxidases (GPXs), and peroxiredoxins (PRXs) [28] (Figure 1, lower panel). The non-enzymatic scavenger glutathione (GSH), also behaves as a “guardian” of redox status, as well as a mediator of redox signal transduction [29] and some reports point to GSH depletion as the main culprits of oxidative stress favoring senescence [30][31]. By maintaining GSH levels, the nuclear factor erythroid 2-related factor 2 (NRF2) behaves as an endogenous booster of redox homeostasis [32][33][34].
The term “Redox Homeostasis” encompasses a dynamic concept: endogenous ROS/RNS, when produced in a regulated manner, are essential signaling molecules conferring outputs for living organisms, and thus their levels are finely regulated under physiological conditions. In fact, cells preserve physiological levels of ROS/RNS as normal oxidant cues (called eustress), on the other hand, cells neutralize ROS/RNS through intracellular defense systems to avoid oxidative challenges (termed distress) [22][35]. Accordingly, extern or internal conditions interrupting redox homeostasis can foster a cell switch into senescence and senescent cells fail to balance their intracellular redox status, skewing towards increased ROS/RNS that fuel senescence signaling and stabilization [4][11][23][36].
Mounting evidence links “regulatory” ncRNAs with redox homeostasis, and thus with the control of the senescence program (discussed in the next section).
3. MiRNAs, lncRNAs and circRNAs in Signaling Pathways of Oxidative stress-induced Senescence (p53, p16, NF-κB, AMPK, SIRT, FOXO, NRF2)
Loss of prooxidant–antioxidant balance, triggered by increased ROS/RNS production or inefficient antioxidant defense systems (primarily due to NRF2 decline) provokes oxidative damages to biomolecules disturbing specific functions, signaling and/or metabolic pathways. This directly or indirectly, through a large set of molecular players, initiates and delivers signaling cascades leading to cell senescence (Figure 2).
Discoveries over the last decades have highlighted that regulatory ncRNAs especially microRNAs, are highly responsive to different types of stimuli, and hence they are implicated in numerous cellular responses, including those involved in oxidative stress-induced senescence [13][17][18][19][20]. By targeting crucial players of antioxidant responses and/or molecular effectors of cell senescence, they can indeed modulate senescence program (Figure 2).
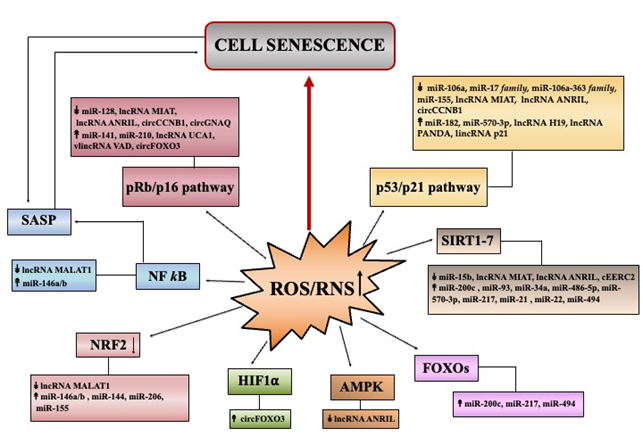
Figure 2. Summary of major pathways initiating senescence through ROS/RNS deregulation. The Figure reports the principal pathways/effectors implicated in redox control of senescence: p53, p16 (cell cycle arrest); NF-κB (SASP profile); AMPK, SIRT, FOXO (metabolic signaling); NRF2 (antioxidant signaling). miRNAs, lncRNAs and circRNAs, found deregulated in senescence, that can modulate specific signaling pathways are indicated. Up and down arrows indicate ncRNA levels that have been found as increased or decreased, respectively in various models of oxidative stress-induced senescence.
The most extensively studied regulatory ncRNAs are microRNAs (miRNAs). They function with a sequence-specific silencing mechanism to guide post-transcriptional repression of targeted transcripts. miRNAs recognize complementary sequences generally localized in the 3’ untranslated regions (3’UTRs) of targeted mRNAs and exhibit conserved sites across mammalian species [37][38][39][40]. miRNAs are essential regulators of most physiological processes of organisms, including differentiation, development, aging, as well as metabolism [41][42] and can influence signaling networks for a variety of stress responses, including those evoked by redox imbalance, via direct or indirect interactions with specific members of redox-induced pathways [43][44]. Of note, the cell redox status could also regulate the biogenesis of miRNAs, thus suggesting another complex interplay between ROS/RNS amounts and miRNA production/levels [45].
Unlike microRNAs, long non-coding RNAs include multiple RNA species longer than 200 nucleotides, which are not translated into proteins [46][47][48]. They are now considered crucial regulators in the gene expression acting both in nuclear and cytoplasmic compartments, and at different levels. Therefore, lncRNAs show a plethora and diversity of functions. The altered expression of several lncRNAs has been associated with oxidative stress-related conditions, including senescence [13][18][47][48]. However, it is important to note that, among the several hundred lncRNAs currently known, only for a few of them the specific functions in oxidative stress response/s have been deeply studied.
The role of circRNAs in oxidative stress-induced senescence is underexplored. The circRNAs are a type of endogenous non-coding RNA molecules that are covalently close and mainly produced by a non-canonical splicing event called “back-splicing” [49][50]. Most of circRNAs act by sequestering miRNAs (miRNA sponges/decoys), while others can interact with RNA-binding proteins to suppress or enhance their activities. In addition, they can function as scaffolds for complex assembly and localization [50].
3.1 Cell Cycle Arrest and SASP Pathways
DNA damage plays a critical role in ROS/RNS-induced premature senescence [5][13]. In particular, the double-strand DNA breaks initiate a DNA damage response (DDR), which if it becomes persistent converges on two DDR effectors: p53/p21 [7,51] and/or pRb/p16 [7][52] (Figure 2). The oncosoppressor protein p53 arrests cell proliferation by inducing the expression of p21Cip1, a member of the cyclin-dependent kinase (CDK) inhibitor family. The pRb is a protein that in an hypophosphorylated state blocks proliferation by sequestering E2F/DP complexes necessary for cell cycle progression. The p16 is an inhibitor of other CDKs and, by preventing CDK-mediated phosphorylation of pRb, favors cell cycle arrest. Several non-coding RNAs act as modulators in p53/p21 and pRb/p16 pathways (Figure 2) .
The first observation that miRNAs can be involved in cell senescence induced by H2O2 comes from the study by Li et al. [53]. The authors demonstrated that in primary human cells, oxidative stress-induced senescence was associated to miRNA changes, with most as downregulated. Further analyses showed that the downregulation of miR-106a, a member of the miR-17 family, is involved in a p53-independent increase of p21, as miR-106a directly targets the p21 3′UTR transcript. miR-182 was found as induced in primary epithelial cells upon H2O2 exposure and its ectopic expression triggers cell senescence through the p53/p21 pathway [54]. Hackl et al. reported that four miRNAs (miR-17, -19b, -20a, -106a) belonging to the miR-17 family and to the paralogous cluster miR-106a-363, were critically expressed at low levels both in cell and organismal aging models [55]. The downregulation of miR-106a-5p and miR-20b-5p was recently confirmed in three human multipotent stromal cell lines treated with H2O2 where they directly regulate specific genes of the p21/CDK/E2F pathway [56]. Another miRNA that has been correlated with oxidative stress-induced senescence is miR-155 [57][58]. In fact, decreased levels of miR-155 caused increase of TP53INP1 promoting p53-mediated growth arrest pathway in WI-38 cells [58]. Baker et al., found that miR-570–3p was highly expressed in COPD (chronic obstructive pulmonary disease), a lung condition characterized by increased oxidative stress and accelerated senescence. They also showed that H2O2-induced expression of miR-570–3p in airway epithelial cells increased p21 levels through the p38MAPK/AP-1 signaling [59] (Figure 2).
Zhuang et al. demonstrated that the increase of the long non-coding RNA H19 (lncH19) enhanced the p53/p21 pathway by scavenging miR-19a that consequently upregulated the suppressor of cytokine signaling 1 (SOCS1) expression (a target of this miRNA) [60]. Other lncRNAs, which contribute to premature senescence by regulating p53/p21 and/or pRb/p16 pathways are lncMIAT [61], lncANRIL [62], lncRNA PANDA [63], lincRNA p21 [64] and VAD [65].
Among circRNAs, Yu et al., found that circCCNB1 was strongly under-expressed in premature senescence of 2BS fibroblasts and its ectopic reduction induced p53, p21, and p16 protein levels [66]. Very recently, Wu et al. reported that silencing of circGNAQ accelerated senescence of endothelial cells and induced p16 protein [67]. Kumar et al. showed that lncUCA1 sequesters hnRNPA1, a RNA-binding protein destabilizing p16INK4a transcripts, thus contributing to improve p16 levels during senescence [68]. Du et al., demonstrated that circular RNA FOXO3 (circFOXO3), increased upon H2O2 treatments, trapped E2F1, HIF1α, and FAK proteins in the cytoplasm, thus inhibiting their functions and promoting senescence [69] (Figure 2).
In parallel to cell cycle arrest, senescent cells express an altered secretory phenotype called senescence-associated secretory phenotype (SASP), containing inflammatory chemokines and cytokines, growth factors, and proteases [13][51][70]. The activation of redox-sensitive NF-κB signaling cascade drives the production of SASP and Schreck et al. first showed that H2O2 mediates the activation of NF-κB transcription factor [71]. NF-κB can be activated by various senescence-inducing pathways and inhibition of canonical NF-κB (RelA/p50) pathway delayed cellular senescence [72].
It has been demonstrated that the activity of NF-κB was determinant for the induction of miR-146a and miR-146b [73], two microRNAs upregulated in H2O2-induced senescence of HCA2 fibroblasts [74]. miR-146a/b by downregulating IRAK, act as negative regulators of pro-inflammatory pathways driven by NF-κB [74]. A possible links of long non-coding RNAs with oxidative-degenerative conditions could be mediated by lncMALAT1 that can intercept the NF-κB/iNOS pathway [75] and/or the p38MAPK signaling cascade [76] (Figure 2).
3.2 Metabolic Pathways
Dysfunctional mitochondria contribute to impaired redox homeostasis and hence senescence. In addition, mounting evidence suggests that beyond ROS/RNS increase, mitochondria can also trigger senescence through multiple cytosolic effectors/pathways [77].
Bioenergetic imbalance can stimulate AMP-activated protein kinase (AMPK) that induces permanent arrest via increased p53/p21 in primary fibroblasts [78] and via pRb/p16 in endothelial cells [79]. Du et al. demonstrated that lncANRIL overexpression alleviated H2O2-induced cell injury by mediating miR-21 upregulation that activates the antioxidant AMPK/β-catenin pathway [80] (Figure 2).
Several studies have shown that if NAD+ declines, the activities of deacetylase sirtuin (SIRT1–7) family are consequently inhibited and this can promote cell senescence [81]. Beyond deacetylation of histones, SIRT1 deacetylates numerous transcription factors, including p53, PPARγ coactivator 1 alpha (PGC-1α), forkhead box O (FOXO), NF-κB, and hypoxia-inducible factor 1 (HIF-1α). By deacetylating p53, SIRT1 suppresses its transcriptional activity, and thus the low NAD+ can induce senescence sustained by the p53/p21 pathway [82]. Several non-coding RNAs are associated to sirtuin-mediated pathway. Lang et al. recently demonstrated that miR-15b directly targets the 3′UTR of mitochondrial SIRT4 and its enforced inhibition boost mitochondrial dysfunctions via increased SIRT4 [83]. Furthermore, Carlomosti et al. [84] demonstrated that upon H2O2 exposure, induction of miR-200c inversely correlates with SIRT1, FOXO1, and eNOS expression, which were validated as its direct targets. Moreover, SIRT1/6 transcripts are direct targets of miR-34a, thus underlying the key role of miR-34a in senescence/aging [85]. Kim et al. demonstrated that miR-486–5p induces premature senescence in human adipose-derived stem cells by inhibiting SIRT1 [86]. Other microRNAs that directly target SIRT1 and that can sustain senescence are: miR-93 [87], miR-217 [88], miR-21 [89], miR-22 [90] and miR-494 [91] (Figure 2).
Regarding the long non-coding RNAs involved in senescence, lncMIAT and lncANRIL have been related to SIRT1 levels [61][62]. They affect the p53 and p16 signaling by sponging miR-181a (ANRIL) and miR-22–3p (MIAT), which directly target SIRT1 transcripts. Similarly, circular RNA EERC2 can contribute to senescence sequestering miR-182–5p that also targets SIRT1 [92] (Figure 2).
FOXO transcription factors are involved in a plethora of biological functions, and thus they can affect senescence [77]. NAD+/sirtuin pathway post-translationally regulates FOXO activities that at least in many long-lived mutants, are causally involved in lifespan by modulating retrograde ROS signaling [77]. FOXO1/3 factors increase catalase and SOD2 expression, hence their inhibition by AKT/PKB (protein kinase B), gives rise to oxidative stress-induced senescence [93]. On the contrary, FOXO4 fosters senescence via p21 increase, and is stimulated through the c-Jun N-terminal kinase (JNK) signaling [94]. Another member of the FOX family, FOXM1, can counteract oxidative stress-induced senescence being involved in the regulation of B cell-specific Moloney murine leukemia virus integration site 1 (BMI-1) expression [95].
The upregulation of miR-200c [84], miR-217 [96] and miR-494 [97] that directly target FOXO transcripts, can be causally involved in ROS/RNS deregulation and oxidative stress-induced senescence (Figure 2).
3.3 NRF2 Antioxidant Pathways
An endogenous booster of redox homeostasis is the nuclear factor erythroid 2-related factor 2 (NRF2) [32][33][34]. NRF2 reshapes antioxidant defense systems by inducing the expression of genes sustaining glutathione levels and metabolism. According to its role, NRF2 amounts are reduced in senescent cells, as well as in aged tissues [98] thereby establishing persistent ROS/RNS production contributing to induction and maintenance of senescence [32] (Figure 2).
Mounting evidence links non-coding RNAs with redox homeostasis and NRF2. Sangokoya et al. reported a functional link between miR-144 and NRF2 levels, since the 3′UTR of NRF2 mRNA can be directly targeted by the miR-144 [99]. Thus, deregulation of miR-144/NRF2 regulatory axis disturbs the redox balance and accordingly, miR-144 was found as upregulated in aged monkey muscle and its expression can be reversed by caloric restriction [100]. Moreover, miR-146a increase was shown to be associated with the decline of NRF2 during rat liver aging [101]. NRF2 can down-regulate expression of miR-206 and this miRNA was indeed found elevated in aged mouse muscle [102], suggesting an association of NRF2 decline with miR-206 increase during aging. Another study demonstrated that miR-155 was increased in the bone marrow of aged mice and pushed ROS generation by suppressing the transcription factor C/EBPβ, which is involved in the regulation of NRF2, SOD1, and HMOX1 expression both in mouse and human MSCs [103].
The lncMALAT1 has been linked to the protective NRF2 signaling in HUVECs during oxidative stress induced by H2O2 [104]. MALAT1 overexpression reduced KEAP1 protein, a negative regulator of NRF2, thus favoring nuclear NRF2 accumulation. This could be mediated by the methyltransferase EZH2, which affects the histone methylation status inhibiting KEAP1 transcription [105] (Figure 2).
4. MiRNAs, lncRNAs and circRNAs in Epigenetic Changes of Oxidative stress-induced Senescence
ROS/RNS imbalance can produce an alteration in the epigenetic landscape directly or indirectly and ROS/RNS amounts could be regulated by epigenetic mechanisms. Epigenetic modifications include DNA methylation, histone modifications, and non-coding RNA activities, which can also work together to sustain the complex program of senescence [106].
4.1 ROS/RNS and DNA Methylation
ROS and RNS can influence the status of DNA methylation [107] (Figure 3). ROS can dampen cytosine methylation status directly by influencing the enzymatic activities of DNA methyltransferases (DNMTs), involved in DNA methylation, while the RNS mechanism is less elucidated. It has been reported that H2O2-induced senescence in normal human cells from different origins is accompanied by decreased expression of DNMT1, thus fostering demethylation of p16INK4a promoter region with consequent expression of p16 [108]. Recently, miR-217 has been causally implicated in DNMT1 decrease in passage-aged human skin fibroblasts [89]; miR-21 expression levels that were upregulated in endothelial cells also targets DNMT1 [89]. Jung et al. reported that miR-29a-3p along with miR-30c-5p directly repressed DNMT3A expression [109]. Accordingly, increased DNMT3 activity due to the decrease of miR-29a-3p and miR-30c-5p impacts negatively on mitochondrial SOD2 expression, and consequently, induces senescence through ROS elevation [109] (Figure 3).
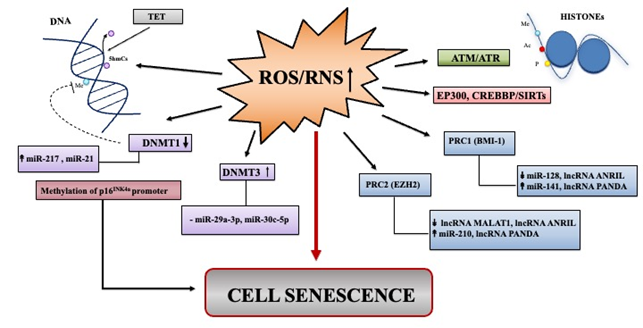
Figure 3. Summary of major epigenetic landscape linked to ROS/RNS induced senescence. Overview of non-coding RNAs deregulated in senescence that can impact epigenetic landscape through ROS/RNS signaling pathways. Up and down arrows indicate ncRNA levels that have been found as increased or decreased, respectively in various models of oxidative stress-induced senescence.
4.2 ROS/RNS and Histone Modifications
ROS/RNS can also increase or decrease histone modifications, such as acetylation and methylation by affecting enzymatic reactions catalyzed by the histone acetylases/deacetylases (HATs/HDACs) and by histone methylases/demethylases (HMT/HDM), respectively. Histone acetylation/deacetylation processes could be shaped by ROS/RNS at various levels, directly or indirectly[107]. Modifications are principally ensured by two cooperating Polycomb group (PcG) complexes: PRC1 (formed by BMI-1, CBX, HPH, RING1) and PRC2 (composed of EED, SUZ12, EZH2, the methyltransferase). In line with this, EZH2 and BMI-1, as well as other components of the PcG pathway become downregulated in senescence prompting changes in chromatin landscape, increased p16INK4a expression, and establishment of the SASP [13][51][70] (Figure 3).
The expression of ncRNAs can influence epigenetic modifications by acting on the members of PRC1 and PRC2, such as BMI-1 and/or EZH2. miR-128, one of the first miRNAs implicated in oxidative stress-induced senescence, directly targets 3′UTR of BMI-1 in medulloblastoma cells [110]. Moreover, the forced expression of miR-141, that also targets BMI-1, blocks proliferation and induces senescence [111]. In searching for miRNAs linked to p16-mediated senescence of human normal mammary epithelial cells, Overhoff et al. found that miR-210 directly targets EED, EZH2, and SUZ12, that consequently mediated the epigenetic increase of p16 [112].
The lncANRIL can inhibits senescence by directly recruit PRC1 and PRC2 to repress p15INK4b, p14ARF, and p16INK4a, respectively [113]. Finally, lncRNA PANDA is able to recruit PRC1/2 components on the genes that activate senescence, such as p21 [63] (Figure 3).

