 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrzej Kasperski | -- | 3155 | 2022-04-14 17:11:23 | | | |
| 2 | Jason Zhu | -37 word(s) | 3118 | 2022-04-15 03:37:24 | | | | |
| 3 | Andrzej Kasperski | + 6 word(s) | 3124 | 2022-04-20 10:22:28 | | |
Video Upload Options
As a phenomenon, cancer is a disease related to multicellular evolution, i.e., cancer in general is understood to be a failure of the multicellular systems and is considered a reversal to unicellularity. Cancer cells are like unicellular organisms that benefit from ancestral-like traits. As a disease, cancer can be interpreted as (a) a destruction of cooperative behaviors underlying multicellular evolution, (b) a disruption of molecular networks established during the emergence of multicellularity or (c) an atavistic state resulting from reactivation of primitive programs typical of the earliest unicellular species. From this point of view and in accordance with the layered model of evolution of cellular functionalities, cancer transformation can occur as a result of huge disturbances or the destruction of functionalities that are located in the multicellular layer.
1. Introduction
2. Molecular Fundamentals of Unified Cell Bioenergetics and Bioenergetic Disturbances
3. Layered Model of Evolution of Cellular Functionalities
3.1. Layer of Cell Bioenergetic Functionalities, i.e., a Phylogenetic Memory Layer of Universal Cell Bioenergetic Functionalities
3.2. Layer of Unicellular Functionalities, i.e., a Phylogenetic Memory Layer of Atavistic Functionalities
3.3. Layer of Multicellular Functionalities, i.e., a Phylogenetic Memory Layer of Multicellular Advanced Functionalities
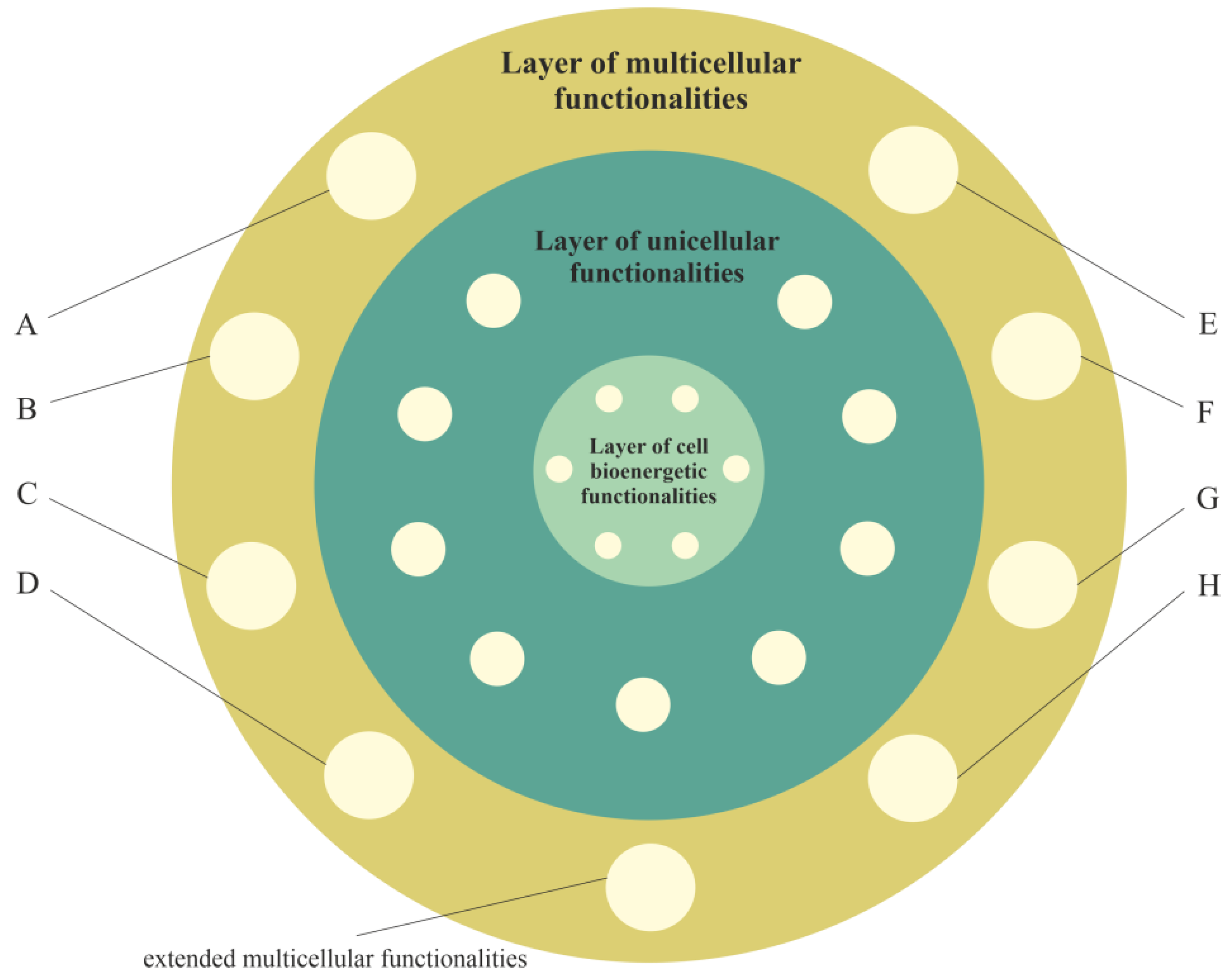
3.4. Cancer Transformation as a Loss of Control over Atavistic Functionalities
Functionalities of the multicellular layer are located in the most external layer of the layered model of evolution of cellular functionalities. In accordance with unified cell bioenergetics, bioenergetic cell problems, especially overenergization of mitochondria, can lead to cancer transformation [30][33]. Cell overenergization (and the related increase in ROS) is followed by adaptation of multiple metabolic strategies to solve this bioenergetic problem [46]. As a result, in light of the layered model of evolution of cellular functionalities, the cell’s response to a huge bioenergetic problem related to overenergization is propagation of disturbances in genome expression from the most internal (i.e., from the layer of bioenergetic functionalities) toward the more external layer (i.e., toward the layer of multicellular functionalities). Functionalities that are localized in the layer of multicellular functionalities, as the most complex and evolutionarily youngest (see Introduction), are the most sensitive to disturbances. The disturbance (or destruction, for example by, high ROS levels) of multicellular layer functionalities can result in a loss of control over functionalities of the unicellular layer, leading to cancer transformation, i.e., uncontrolled activity of atavistic functionalities (Figure 2).
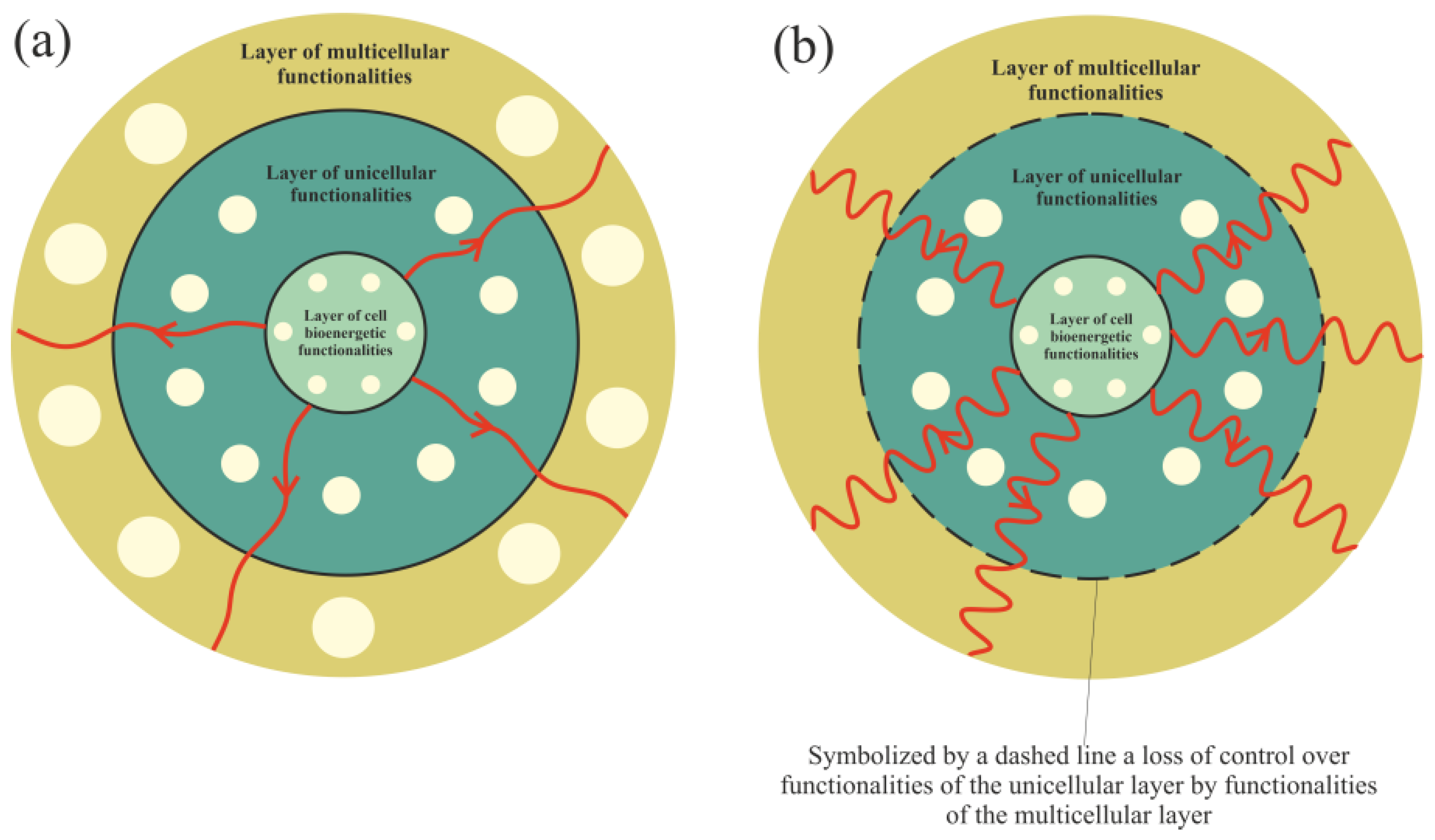
Figure 2. Propagation of disturbances in cell bioenergetics. (a) – Small disturbances cause small disturbances in functionalities of the unicellular and multicellular layers. (b) – Huge disturbances can cause a loss of functionalities of the multicellular layer (or huge disturbances in their operation), leading to a loss of control over unicellular layer functionalities.
This entry is adapted from 10.3390/ijms23074017
References
- Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101.
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2020; in press.
- Heng, H.H. Debating Cancer: The Paradox in Cancer Research; World Scientific Publishing Co.: Singapore, 2015; ISBN 978-981-4520-84-3.
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162.
- Baker, S.G. The detached pericyte hypothesis: A novel explanation for many puzzling aspects of tumorigenesis. Org. J. Biol. Sci. 2018, 2, 25–41.
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880.
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542.
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell. Sci. 2003, 116, 4095–4106.
- Baker, S.G. Paradoxes in carcinogenesis should spur new avenues of research: An historical perspective. Disrupt. Sci. Technol. 2012, 1, 100–107.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W., Jr. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Baker, S.G. The case for a cancer paradox initiative. Carcinogenesis 2021, 42, 1023–1025.
- Arguello, F. Atavistic Metamorphosis: A New and Logical Explanation for the Origin and Biological Nature of Cancer: With a Discussion on a Novel Approach to Treat Cancer; CreateSpace: Scotts Valley, CA, USA, 2011; ISBN 13:978-1460968994.
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. World 2013, 26, 37–40.
- Davies, P.C.W.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 15001.
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20.
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92.
- Erenpreisa, J.; Cragg, M.S. Life-cycle features of tumour cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71.
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018.
- Erenpreisa, J.; Wheatley, D. Endopolyploidy in development and cancer; “survival of the fattest?”. Cell Biol. Int. 2005, 29, 981–982.
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. BioEssays 2014, 36, 827–835.
- Niculescu, V.F. Developmental and non developmental polyploidy in xenic and axenic cultured stem cell lines of Entamoeba invadens and E. histolytica. Insights Stem. Cells 2016, 2, 1–9.
- Vincent, M.D. Cancer: A de-repression of a default survival program common to all cells? BioEssays 2012, 34, 72–82.
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350.
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, 2000305.
- Yett, J.R. Eukaryotes. In Salem Press Encyclopedia of Science; Grey House Publishing: Amenia, NY, USA, 2015.
- Khozouz, R. Atavistic cancermodel: A new theory of cancer? BioEssays 2021, 43, 2100206.
- Cui, Q.; Wen, S.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach: Recent updates. Future Med. Chem. 2017, 9, 929–949.
- Cross, W.; Kovac, M.; Mustonen, V.; Temko, D.; Davis, H.; Baker, A.; Biswas, S.; Arnold, R.; Chegwidden, L.; Gatenbee, C.; et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018, 2, 1661–1672.
- Regoes, R.R. Population genetics meets cancer genomics. Proc. Natl. Acad. Sci. USA 2010, 107, 18241–18242.
- Kasperski, A.; Kasperska, R. Bioenergetics of life, disease and death phenomena. Theory Biosci. 2018, 137, 155–168.
- Kasperski, A. Modelling of cells bioenergetics. Acta Biotheor. 2008, 56, 233–247.
- Noda, M.; Yamashita, S.; Takahashi, N.; Eto, K.; Shen, L.M.; Izumi, K.; Daniel, S.; Tsubamoto, Y.; Nemoto, T.; Iino, M.; et al. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J. Biol. Chem. 2002, 277, 41817–41826.
- Kasperski, A.; Kasperska, R. Study on attractors during organism evolution. Sci. Rep. 2021, 11, 9637.
- Kasperski, A. Genome Attractors as Places of Evolution and Oases of Life. Processes 2021, 9, 1646.
- Kasperski, A.; Kasperska, R. Selected disease fundamentals based on the unified cell bioenergetics. J. Investig. Biochem. 2013, 2, 93–100.
- Bakker, B.M.; Overkamp, K.M.; van Maris, A.J.; Kötter, P.; Luttik, M.A.; van Dijken, J.P.; Pronk, J.T. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 15–37.
- Overkamp, K.M.; Bakker, B.M.; Kötter, P.; van Tuijl, A.; de Vries, S.; van Dijken, J.P.; Pronk, J.T. In vivo analysis of the mechanisms for oxidation of cytosolic NADH by Saccharomyces cerevisiae mitochondria. J. Bacteriol. 2000, 182, 2823–2830.
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004, 87, 2060–2073.
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanizm. J. Cell. Biol. 2006, 175, 913–923.
- Yu, Q.; Heikal, A.A. Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobio. B Biol. 2009, 95, 46–57.
- Mukhopadhyay, A.; Bose, C.K. Debating cancer: The paradox in cancer research. Indian J. Med. Res. 2017, 146, 435–436.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591.
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545.
- Alfarouk, K.O.; Shayoub, M.E.; Muddathir, A.K.; Elhassan, G.O.; Bashir, A.H. Evolution of Tumor Metabolism might Reflect Carcinogenesis as a Reverse Evolution process (Dismantling of Multicellularity). Cancers 2011, 3, 3002–3017.
- Diaz-Ruiz, R.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim. Biophys. Acta 2009, 1796, 252–265.
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870.
- Lu, W.; Ogasawara, M.A.; Huang, P. Models of reactive oxygen species in cancer. Drug Discov. Today Dis. Models 2007, 4, 67–73.
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci. 2009, 66, 3663–3673.
- Wen, S.; Zhu, D.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med. Chem. 2013, 5, 53–67.
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692.
- Aktipis, C.A.; Nesse, R.M. Evolutionary foundations for cancer biology. Evol. Appl. 2013, 6, 144–159.
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer adaptations: Atavism, de novo selection, or something in between? BioEssays 2017, 39, 1700039.
- Mazzocca, A. The Systemic–Evolutionary Theory of the Origin of Cancer (SETOC): A New Interpretative Model of Cancer as a Complex Biological System. Int. J. Mol. Sci. 2019, 20, 4885.
- Kováč, L. Lamarck and Darwin revisited. EMBO Rep. 2019, 20, e47922.
- Lineweaver, C.H.; Davies, P.C.W. Comparison of the Atavistic Model of Cancer to Somatic Mutation Theory: Phylostratigraphic Analyses Support the Atavistic Model. In The Physics of Cancer Research Advances; Gerstman, B.S., Ed.; World Scientific: Singapore, 2020; Volume 12, pp. 243–261.
- Cornish-Bowden, A. Lynn Margulis and the origin of the eukaryotes. J. Theor. Biol. 2017, 434, 1.
- Grosberg, R.K.; Strathmann, R.R. The Evolution of Multicellularity: A Minor Major Transition? Annu. Rev. Ecol. Evol. Syst. 2007, 38, 621–654.
- Nedelcu, A.M. The evolution of multicellularity and cancer: Views and paradigms. Biochem. Soc. Trans. 2020, 48, 1505–1518.


