Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marta Borges Canha | -- | 3304 | 2022-04-14 14:25:58 | | | |
| 2 | Rita Xu | Meta information modification | 3304 | 2022-04-15 03:57:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Canha, M.; , .; Neves, J.S.; Carvalho, D.; Leite Moreira, A. Nonalcoholic Fatty Liver Disease and Endocrine Axes. Encyclopedia. Available online: https://encyclopedia.pub/entry/21773 (accessed on 21 June 2026).
Canha M, , Neves JS, Carvalho D, Leite Moreira A. Nonalcoholic Fatty Liver Disease and Endocrine Axes. Encyclopedia. Available at: https://encyclopedia.pub/entry/21773. Accessed June 21, 2026.
Canha, Marta, , João Sérgio Neves, Davide Carvalho, Adelino Leite Moreira. "Nonalcoholic Fatty Liver Disease and Endocrine Axes" Encyclopedia, https://encyclopedia.pub/entry/21773 (accessed June 21, 2026).
Canha, M., , ., Neves, J.S., Carvalho, D., & Leite Moreira, A. (2022, April 14). Nonalcoholic Fatty Liver Disease and Endocrine Axes. In Encyclopedia. https://encyclopedia.pub/entry/21773
Canha, Marta, et al. "Nonalcoholic Fatty Liver Disease and Endocrine Axes." Encyclopedia. Web. 14 April, 2022.
Copy Citation
Nonalcoholic fatty liver disease (NAFLD) is the leading cause of chronic liver disease. NAFLD often occurs associated with endocrinopathies. Evidence suggests that endocrine dysfunction may play an important role in NAFLD development, progression, and severity.
NAFLD
fatty liver
lipidomics
metabolic modeling
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the leading cause of chronic liver disease. NAFLD is a metabolic liver disease that encompasses a wide spectrum from simple steatosis to steatohepatitis (NASH) and fibrosis to cirrhosis and hepatocarcinoma [1]. It has also been termed a “barometer of metabolic health” due to its metabolic roots [2]. A group of experts on the theme have recently reached a consensus, saying that this entity might be known as MAFLD (metabolic-associated fatty liver disease) and be diagnosed by positive criteria rather than being an exclusion diagnosis (requiring the exclusion of other causes chronic liver diseases before diagnosing NAFLD) [3]. Its high prevalence, complex pathogenesis, and lack of approved therapies make this disease a hot topic of scientific research [4].
NAFLD often occurs associated with endocrinopathies, as it has been increasingly recognized [5][6]. Digging into these associations may improve the current knowledge on this disease [7]. Moreover, emerging evidence suggests that endocrine dysfunction may play an important role in NAFLD development, progression, and severity [8]. Moreover, a variety of rare hereditary liver and intestinal diseases as well as several drugs may trigger or worsen NAFLD; this further highlights the complexity in understanding NAFLD [9].
2. Hypothalamic and Pituitary Dysfunction
2.1. Growth Hormone (GH)
2.1.1. Adult GH Deficiency
While essential for linear growth during childhood, GH promotes lipolysis, especially in visceral adipose tissue and protein synthesis and decreases peripheral insulin sensitivity and glucose uptake in adults [10]. Adult GH deficiency is a clinical syndrome that mainly results from pituitary tumors or the treatment of such tumors, namely, with surgery or radiation. Regardless of etiology, GH deficiency is generally associated with several metabolic changes, including increased visceral adipose tissue, decreased lean body mass, and dyslipidemia, with higher total and low-density lipoprotein (LDL) cholesterol, triglycerides, and hypertension [11][12]. Although GH has antagonistic effects on insulin action, its deficiency leads to insulin resistance and glucose intolerance, probably due to specific changes in fat distribution, such as increased visceral adipose tissue and ectopic fat accumulation [10][13]. These frequently lead to metabolic syndrome (MS) in patients with untreated GH deficiency [13]. Given the intricate association between MS and NAFLD, a focus has been given to the role of GH in the pathogenesis of NAFLD in the last decade (Figure 1).
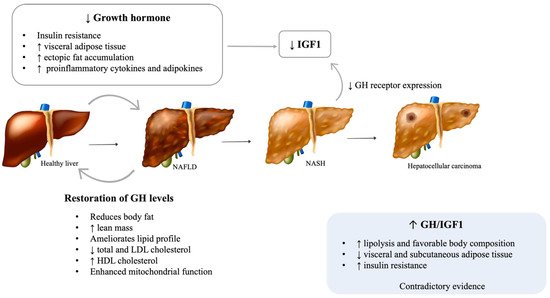
Figure 1. Possible mechanisms of GH deficiency on NALFD development and progression. NAFLD: Nonalcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis; GH: Growth hormone; IGF1: Insulin-like growth factor 1. [Viktoriia Kasyanyuk] © https://www.123rf.com/ (accessed on 22 February 2022) and Servier Medical Art by Servier.
Several cross-sectional studies reported an increased prevalence of liver dysfunction and NAFLD in patients with hypopituitarism, particularly those with GH deficiency [14][15][16]. Moreover, patients with GH deficiency and NAFLD have an accelerated progression of the hepatic disease [15]. Additionally, a study by Koehler et al. showed that obese patients with NASH and advanced fibrosis have low serum GH levels and that normal GH levels essentially excluded advanced fibrosis [17][18]. Patients with Laron syndrome, which is characterized by GH resistance due to inactivating mutations of the GH receptor, were also found to have a higher incidence of NAFLD [19]. Interestingly, acromegalic patients treated with the GHR antagonist pegvisomant showed increases in hepatic triglyceride content [20]. Despite the aforementioned results, evidence on this matter remains conflicting, with some studies not finding differences in the prevalence of NAFLD or in the intrahepatic lipid content assessed by magnetic resonance spectroscopy between GH deficient patients and healthy adults [21]. Heterogeneity in sample sizes and the clinical characteristics of included patients, such as sex and ethnicity, may help to explain these differences in the studies’ results.
Animal and cell culture studies further support a role for the GH/ insulin-like growth factor-1 (IGF-1) axis in the pathophysiology of NAFLD and its progression to NASH and fibrosis [11]. Animal models with liver-specific mutations in the GH receptor or downstream signaling pathways (JAK2/STAT5) develop metabolic syndrome, hepatic steatosis, steatohepatitis, and fibrosis [22][23][24]. Moreover, in animal models of adult-onset hepatic GH resistance, steatosis and NASH evolve rapidly after the loss of hepatic GH signaling, regardless of other signs of metabolic dysfunction [25].
The restoration of GH levels in adults with GH deficiency reduces body fat, increases lean mass, and ameliorates the lipid profile, decreasing total and LDL cholesterol and increasing high-density lipoprotein (HDL) cholesterol [26][27]. A few small studies have shown that GH replacement improves hepatic injury, as observed by a rapid decrease in serum liver transaminases and gamma-glutamyl transferase levels, steatosis, lobular inflammation, hepatocyte ballooning, and the severity of fibrosis [15][28]. Additionally, GH supplementation in pediatric patients with GH deficiency has been associated with an improvement in NAFLD, as it decreases visceral fat accumulation and lipid deposition on the liver and enhances mitochondrial function [29][30][31]. Animal studies have reinforced these results [24]. It is well known that the actions of GH are mediated both directly and indirectly through the stimulation of IGF-1 production [10]., and the majority of circulating IGF-1 (>90%) is produced by hepatocytes in response to growth hormone receptor stimulation [32]. Increasing evidence suggests that both GH and IGF-1 have direct and indirect effects on hepatic structure and function [33]. Moreover, decreased GH and, consequently, IGF-1 might be responsible for the muscle mass changes, particularly sarcopenia, which is seen in NAFLD [34]. GH was recently proposed to directly inhibit de novo lipogenesis and the expression of peroxisome proliferator-activated receptor-gamma (PPAR-γ) and CD36, key regulators of free fatty acid uptake [23][25]. IGF-1 is an anti-inflammatory molecule that contributes to mitochondrial function and reduces oxidative stress in the liver [24]. IGF-1 prevents cholesterol accumulation through the stimulation of the expression of ATP-binding cassette transporter A1 (ABCA1), a pivotal regulator of lipid efflux from cells to apolipoproteins [35]. Furthermore, IGF-1 limits the activity of hepatic stellate cells and induces their senescence, therefore attenuating hepatic fibrosis [36]. Lower levels of IGF-1 might result in lower protection against liver inflammation and fibrosis [37]. By impairing the adipose tissue phenotype, GH deficiency increases the expression of proinflammatory cytokines and adipokines [38], which compromises insulin sensitivity and impairs the ability of adipose tissue to store fat, increasing lipid influx into ectopic organs, such as the liver [22]. Chronic liver diseases, including NASH, are associated with a reduction of GH receptor (GHR) expression and, therefore, reduced IGF-1 levels [39]. As described above, lower levels of IGF-1 impair liver homeostasis, with a higher risk of fibrosis, leading to a vicious cycle of both worsening hepatic homeostasis and increasing growth hormone function.
Given the crucial role of GH in hepatic lipid metabolism, there are some clinical trials examining the impact of low-dose GH supplementation in patients with hepatic steatosis and NASH without known hypothalamic/pituitary disease. A new clinical trial had its results recently published, showing that treatment with recombinant human GH may have the potential to reduce liver fat content in adolescents with NAFLD and obesity [40]. Other clinical trials studying the impact of GH supplementation on NAFLD are underway, such as the clinical trial named Growth Hormone and Intrahepatic Lipid Content in Patients With Nonalcoholic Fatty Liver Disease (NCT02217345). Lastly, IGF-1 replacement is also being considered as an option to treat patients with liver diseases [41]. Experimental studies show that treatment with IGF-1 is particularly beneficial in the reduction of liver fibrosis, although positive effects in hepatic steatosis and inflammation can also be seen [24][42].
2.1.2. Acromegaly
Acromegaly is characterized by excessive GH and, consequently, IGF-1 and is the most frequent cause of GH-secreting pituitary adenoma. Increased levels of GH are associated with increased lipolysis and favorable body composition, with increased lean body mass and decreased visceral and subcutaneous adipose tissue [43]. However, acromegaly also promotes insulin resistance, with consequent hyperglycemia, hyperinsulinemia, hypertriglyceridemia, and an increased risk of overt diabetes [44]. These paradoxical effects may justify contradictory evidence on this topic. While some studies including patients with active acromegaly found that intrahepatic lipid, measured by magnetic resonance spectroscopy, is relatively low in comparison to healthy subjects [20][45], others showed that hepatic steatosis is a common comorbidity in acromegaly, hypothesizing that lipotoxicity and insulin resistance may outweigh the direct hepatic effects of GH [46].
Acromegaly treatment with surgery or medical therapy improves metabolic risk by increasing insulin sensitivity [45]. However, GH, IGF-1, insulin-like growth factor binding proteins (IGFBPs), and medical treatment have a complex relationship with insulin sensitivity and hepatic steatosis. GHR antagonists (GHRA) induce an improvement in acromegaly glycemic control through the decrease of glucose and the normalization of insulin secretion [47]. This effect enables one to understand the important effect of GH on hepatic and peripheral IGF-1 action. Hepatic GH-induced IGF-1 production is regulated by portal insulin levels, as insulin promotes the translocation of hepatic GHR to the surface. When portal insulin levels are high, the liver becomes GH sensitive, regardless of the cause of the increased production of insulin. In addition, portal insulin also inhibits hepatic IGFBP1 production, which may increase the bioavailability of circulating IGF-1. Insulin suppression by somatostatin analogs (SSA) also selectively results in hepatic GH resistance, which itself decreases hepatic IGF-1 production [48]. Therefore, the consequent reduction in circulating IGF-1 does not necessarily reflect GH activity in peripheral tissues. It, thus, makes sense that the normalization of serum IGF-1 levels during SSA does not necessarily imply the control of disease’s activity in peripheral tissues, which is a condition that Neggers coined as being “extra-hepatic acromegaly” [49]. This concept received support in a study that evaluated surgically and SSA-treated acromegalics. Despite the normalization of IGF-1, SSA-treated patients had less suppressed GH levels and less symptom relief [50]. On the other hand, GHRA do not block all tissues with equal effectiveness for the GH actions. Adipose tissue seems to require less GHRA to reduce GH actions when compared to the liver, where more GHRA are required to reduce IGF-1 production [51]. This could be a reason for local GHRA-induced lipomatosis. In further support of this hypothesis, it was recently reported that short-term GHRA administration in healthy subjects can suppress lipolysis without affecting either circulating or local IGF-1 [52]. Accordingly, it is possible that peripheral suppression of GH activity is obtained prior to the normalization of hepatic IGF-1 production. Therefore, GHRA-treated patients with acromegaly and normal peripheral IGF-1 can have peripheral GH deficiency [49]. If all this is true, then patients with acromegaly and diabetes should need higher GHRA doses to normalize IGF-1 compared to patients without diabetes. Recently, this was demonstrated by Droste et al. [53].
In a cross-sectional study including patients previously treated for acromegaly, hepatic steatosis, measured by magnetic resonance spectroscopy, was found to be increased compared to healthy controls, even several years after successful treatment [54]. In a recent prospective study, Ciresi et al. found no differences in the prevalence of steatosis after 12 months of treatment with somatostatin analogs, measured by abdominal ultrasonography [55]. The heterogeneity in the chosen imaging method to assess hepatic steatosis and in the treatments for acromegaly may explain these differences.
2.2. Hyperprolactinemia
Prolactin is a polypeptide hormone produced by lactotroph cells in the anterior pituitary [56]. Prolactin release is mainly controlled by hypothalamic inhibitory tone through dopamine and the stimulatory influences of thyroid stimulating hormone (TSH)-releasing hormone and circulating estrogens. Its major functions are related to pregnancy and lactation [57]. In addition, prolactin is involved in the regulation of the immune system, food intake, and bone formation [56].
A growing body of evidence supports prolactin as an active contributor to human metabolic health [57]. Namely, experimental animal studies found that it stimulates pancreatic β cell proliferation and insulin gene transcription, modulates lipid metabolism in adipose tissue, and induces adipogenesis [58][59]. Despite the beneficial roles of prolactin on metabolic homeostasis, pathological increases in prolactin levels have been frequently associated with metabolic disturbances, namely, weight gain, obesity, hyperinsulinemia, and reduced insulin sensitivity, all considered important players in the pathogenesis of NAFLD [60]. Normalization of prolactin levels with dopamine agonists correlated with weight loss, although some studies have shown a more pronounced weight loss in men, suggesting a gender difference [61]. Furthermore, treatment with dopamine agonists improves insulin sensitivity, glycemic control, and lipid profile, reducing triglycerides and total and LDL cholesterol [62].
The impact of prolactin on liver function and structure is poorly understood (Figure 2). The presence of functional prolactin receptors in hepatocytes has also been previously demonstrated [63]. Recent in vitro and in vivo studies suggest that prolactin protects the liver against lipid accumulation by decreasing the expression of CD36 and stearoyl-coenzyme A desaturase 1 (SCD1), an enzyme involved in fatty acid biosynthesis [64][65]. In rodents, prolactin is thought to be involved in the regulation of liver insulin sensitivity [66]. In human studies, lower prolactin levels were found in patients with more severe hepatic steatosis, suggesting a possible involvement of prolactin in the progression of this disease [64]. Although prolactin is thought to reduce liver fat content, it is plausible that chronic hyperprolactinemia is involved in the development of NAFLD. Despite the absence of studies evaluating liver function and structure in patients with hyperprolactinemia, a few animal studies support this hypothesis. Luque et al. suggest that prolactin may be directly involved in changes in the signaling pathways of de novo lipogenesis, which lead to fatty liver [67]. In diabetic murine models, the triglyceride content in the liver increased with the administration of high doses of prolactin [68].
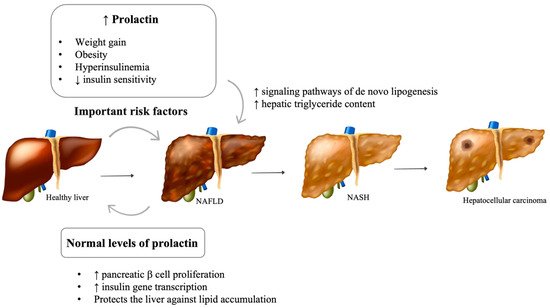
Figure 2. Impact of prolactin levels on NAFLD development and progression. NAFLD: Nonalcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis; GH: Growth hormone; IGF1: Insulin-like growth factor 1. [Viktoriia Kasyanyuk] © https://www.123rf.com/ (accessed on 22 February 2022) and Servier Medical Art by Servier.
2.3. Vasopressin Disturbances
Vasopressin (V), also known as antidiuretic hormone (ADH), has a crucial role in the regulation of water balance, vascular tone, and the endocrine stress response [69]. Due to its short half-life, very low concentration, small size, and poor stability in plasma samples, clinical studies indirectly determine the levels of ADH in the circulation through the measurement of copeptin, which is produced in equimolar quantities with ADH [69].
A role of ADH in the regulation of glucose and lipid metabolism has been acknowledged by several epidemiological and experimental studies [70]. Patients with diabetes mellitus have markedly increased levels of ADH in comparison with healthy subjects [71]. Whether copeptin is predictive of the development of diabetes-induced NAFLD remains unknown. Recent studies demonstrated a strong association between high copeptin levels and the prevalence and severity of both NAFLD and NASH [71][72].
Several ex vivo and in vitro studies have shown that ADH intensifies hyperglycemia. It enhances hepatic gluconeogenesis and glycogenolysis through the activation of V1a receptors [73]. ADH also induces vasoconstriction, contributing to liver hypoxia, and further stimulates glycogenolysis [74]. and stimulates the release of pituitary adrenocorticotropin hormone via V1b receptors, increasing the release of cortisol, which is thought to be an important contributor to ADH-induced hyperglycemia and insulin resistance [75].
Despite being controversial and less understood, most studies suggest that ADH decreases plasma non-esterified fatty acids [76][77]. ADH promotes lipogenesis in the hepatic tissue through the V1a receptor and inhibits lipolysis in adipocytes [76][78]. The V-induced decrease in plasma non-esterified fatty acids may reduce its supply to the liver [77]. On the other hand, one study found the role of vasopressin in fatty acid synthesis and lipogenesis varied with different incubating media of rat hepatocytes, suggesting that vasopressin function may vary according to the nutritional status [79]. Taveau et al. found reduced levels of hepatic cholesterol and triacylglycerol and a lower expression of genes involved in lipogenesis in well-hydrated obese rats with low levels of ADH [80].
3. Phosphocalcic Metabolism Disturbances
3.1. Vitamin D Deficiency
Vitamin D is classically recognized for its role in phosphocalcium metabolism and bone health. Notwithstanding, its receptors are present ubiquitously, and it is associated with several effects in various organs and systems [81][82]. Vitamin D deficiency seems to be associated with several metabolic disturbances [83][84][85], namely, NAFLD, although inconsistently.
Fundamental research studies have shown that vitamin D exerts anti-inflammatory, anti-proliferative, and anti-fibrotic effects in the liver [86]. In vitro and in vivo studies suggest that this protective effect may be partially mediated by stellate cells through the inhibition of fibrogenesis [83][86][87] Using mouse hepatocytes, Dong B. et al. showed that vitamin D receptor activation in macrophages by vitamin D ligands ameliorates liver inflammation and steatosis [88]. Moreover, obese animals with vitamin D deficiency presented with greater NAFLD progression, and vitamin D supplementation improved hepatic morphology and function [83]. Contrariwise, Bozic et al. showed that hepatic vitamin D receptor activation promotes high-fat diet-associated liver steatosis in a mouse model [89].
Human studies are also conflicting. Several epidemiological studies lead towards an association between low vitamin D levels and NAFLD, though no causal relationship has been found [90][91][92]. Namely, Targher et al. showed that patients with biopsy-proven NAFLD had lower vitamin D levels compared to controls [93]. Furthermore, an analysis of data from the National Health and Nutrition Examination Survey (NHANES III) claims that vitamin D levels are inversely associated with the severity of NAFLD [94]. A recent study from researchers group associated vitamin D deficiency with a higher risk of hepatic steatosis in individuals with morbid obesity [95]. An analysis from the Sixth Korea National Health and Nutrition Examination Survey (KNHANES VI) argued against such association [96]. In a Mendelian randomization analysis, Wang et al. found no causal association between vitamin D and NAFLD in a Chinese population with over 9000 participants [97]. Additionally, Barchetta et al. performed a randomized, double-blind, placebo-controlled trial (RCT) and concluded that vitamin D supplementation, in a high oral dose for a period of 24 weeks did not improve hepatic steatosis in patients with NAFLD and type 2 diabetes [98][99][100]. In another RCT in NAFLD patients, no beneficial effects on liver function were seen when comparing supplementation with vitamin D, calcitriol, and placebo [101]. Therefore, no strong recommendations currently exist concerning vitamin D supplementation in patients with NAFLD [102][103].
Pacifico L. et al. dwelled on the possible confounders of a NAFLD/NASH and vitamin D deficiency association, namely, the influence of the host and environment in vitamin D levels as well as the great variability in laboratory methods and intra-individual variability in this vitamin’s level [104]. More studies, particularly well-powered randomized controlled trials, are needed to evaluate the potential role of vitamin D supplementation in the management of NAFLD.
3.2. Other Disturbances of Bone Metabolism
The deterioration of bone homeostasis has been incongruously associated with NAFLD, and the pathophysiology of such association remains to be elucidated [105]. It is hypothesized that very complex mechanisms are involved, and a plausible perspective centralizes the causal pathway in the liver. The dysfunctional visceral adipose tissue that often exists concurrently with NAFLD is a great source of pro-inflammatory, pro-coagulant, and profibrogenic factors that may contribute to this association. The state of insulin resistance commonly present in NAFLD patients may also play an important role as well as the vitamin D deficit that may relate to such hepatic disorders, as previously mentioned [105][106][107].
An observational study in a Chinese population showed that the liver fat content was inversely correlated with bone mineral density (BMD) in middle-aged and elderly men but not in women [108]. Additionally, in a cohort of Chinese participants that were 40 years old or older, the prevalence of osteoporotic fractures was significantly greater in men with NAFLD but not in women [109]. Moreover, a Korean population-based study presented a detrimental effect of NAFLD on BMD in men but an unexpected positive effect in postmenopausal women [110]. There are some studies that show similar harmful effects for both genders (namely postmenopausal women), but others evidence a detrimental effect in postmenopausal women [111][112]. Bhatt et al. stated that PTH levels are independently associated with NAFLD in Asian Indians [113]. Similarly, a metanalysis that included observational studies focusing on bone mineral density claimed that no correlation exists between bone mineral density and NAFLD [114]. It is important to recognise that most of the studies mentioned are in Asian populations, suggesting an ethnic difference in the impact of the phosphocalcic axis on the liver.
Despite being a controversial theme of the debate, it is of great interest to clarify the existence of such links in future prospective studies, namely, addressing gender, ethnic, and age differences. The routine screening of BMD in NAFLD patients may become an important addition to the management of these complex patients.
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Sookoian, S.; Pirola, C.J. Personalizing care for nonalcoholic fatty liver disease patients: What are the research priorities? Pers. Med. 2014, 11, 735–743.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209.
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285.
- Singeap, A.M.; Stanciu, C.; Huiban, L.; Muzica, C.M.; Cuciureanu, T.; Girleanu, I.; Chiriac, S.; Zenovia, S.; Nastasa, R.; Sfarti, C.; et al. Association between Nonalcoholic Fatty Liver Disease and Endocrinopathies: Clinical Implications. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6678142.
- Marino, L.; Jornayvaz, F.R. Endocrine causes of nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 11053–11076.
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324.
- Hazlehurst, J.M.; Tomlinson, J.W. Non-alcoholic fatty liver disease in common endocrine disorders. Eur. J. Endocrinol. 2013, 169, R27–R37.
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and management of secondary causes of steatohepatitis. J. Hepatol. 2021, 74, 1455–1471.
- Kargi, A.Y.; Merriam, G.R. Diagnosis and treatment of growth hormone deficiency in adults. Nat. Rev. Endocrinol. 2013, 9, 335–345.
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447.
- Gazzaruso, C.; Gola, M.; Karamouzis, I.; Giubbini, R.; Giustina, A. Cardiovascular risk in adult patients with growth hormone (GH) deficiency and following substitution with GH--an update. J. Clin. Endocrinol. Metab. 2014, 99, 18–29.
- Thomas, J.D.; Monson, J.P. Adult GH deficiency throughout lifetime. Eur. J. Endocrinol. 2009, 161 (Suppl. 1), S97–S106.
- Nguyen, A.; Ricolfi, F.; Lemogne, B.; Aho, S.; Lemaire, S.; Bouillet, B.; Duvillard, L.; Denimal, D.; Fourmont, C.; Loffroy, R.; et al. Liver Fat Content in People with Pituitary Diseases: Influence of Serum IGF1 Levels. Horm. Metab. Res. 2018, 50, 303–307.
- Nishizawa, H.; Iguchi, G.; Murawaki, A.; Fukuoka, H.; Hayashi, Y.; Kaji, H.; Yamamoto, M.; Suda, K.; Takahashi, M.; Seo, Y.; et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur. J. Endocrinol. 2012, 167, 67–74.
- Ichikawa, T.; Hamasaki, K.; Ishikawa, H.; Ejima, E.; Eguchi, K.; Nakao, K. Non-alcoholic steatohepatitis and hepatic steatosis in patients with adult onset growth hormone deficiency. Gut 2003, 52, 914.
- Koehler, E.; Swain, J.; Sanderson, S.; Krishnan, A.; Watt, K.; Charlton, M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: An endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012, 32, 279–286.
- Lonardo, A.; Loria, P.; Leonardi, F.; Ganazzi, D.; Carulli, N. Growth hormone plasma levels in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2002, 97, 1071–1072.
- Laron, Z.; Ginsberg, S.; Webb, M. Nonalcoholic fatty liver in patients with Laron syndrome and GH gene deletion—Preliminary report. Growth Horm. IGF Res. 2008, 18, 434–438.
- Madsen, M.; Krusenstjerna-Hafstrom, T.; Moller, L.; Christensen, B.; Vendelbo, M.H.; Pedersen, S.B.; Frystyk, J.; Jessen, N.; Hansen, T.K.; Stodkilde-Jorgensen, H.; et al. Fat content in liver and skeletal muscle changes in a reciprocal manner in patients with acromegaly during combination therapy with a somatostatin analog and a GH receptor antagonist: A randomized clinical trial. J. Clin. Endocrinol. Metab. 2012, 97, 1227–1235.
- Gardner, C.J.; Irwin, A.J.; Daousi, C.; McFarlane, I.A.; Joseph, F.; Bell, J.D.; Thomas, E.L.; Adams, V.L.; Kemp, G.J.; Cuthbertson, D.J. Hepatic steatosis, GH deficiency and the effects of GH replacement: A Liverpool magnetic resonance spectroscopy study. Eur. J. Endocrinol. 2012, 166, 993–1002.
- Sos, B.C.; Harris, C.; Nordstrom, S.M.; Tran, J.L.; Balazs, M.; Caplazi, P.; Febbraio, M.; Applegate, M.A.; Wagner, K.U.; Weiss, E.J. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J. Clin. Investig. 2011, 121, 1412–1423.
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011, 152, 181–192.
- Nishizawa, H.; Takahashi, M.; Fukuoka, H.; Iguchi, G.; Kitazawa, R.; Takahashi, Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem. Biophys. Res. Commun. 2012, 423, 295–300.
- Cordoba-Chacon, J.; Majumdar, N.; List, E.O.; Diaz-Ruiz, A.; Frank, S.J.; Manzano, A.; Bartrons, R.; Puchowicz, M.; Kopchick, J.J.; Kineman, R.D. Growth Hormone Inhibits Hepatic De Novo Lipogenesis in Adult Mice. Diabetes 2015, 64, 3093–3103.
- Newman, C.B.; Carmichael, J.D.; Kleinberg, D.L. Effects of low dose versus high dose human growth hormone on body composition and lipids in adults with GH deficiency: A meta-analysis of placebo-controlled randomized trials. Pituitary 2015, 18, 297–305.
- Maison, P.; Griffin, S.; Nicoue-Beglah, M.; Haddad, N.; Balkau, B.; Chanson, P.; Metaanalysis of Blinded, R.P.-C.T. Impact of growth hormone (GH) treatment on cardiovascular risk factors in GH-deficient adults: A Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J. Clin. Endocrinol. Metab. 2004, 89, 2192–2199.
- Takahashi, Y.; Iida, K.; Takahashi, K.; Yoshioka, S.; Fukuoka, H.; Takeno, R.; Imanaka, M.; Nishizawa, H.; Takahashi, M.; Seo, Y.; et al. Growth hormone reverses nonalcoholic steatohepatitis in a patient with adult growth hormone deficiency. Gastroenterology 2007, 132, 938–943.
- Miranda-Lora, A.L.; Zamora-Nava, L.E.; Marin-Rosas, D.L.; Klunder-Klunder, M.; Sanchez-Curiel, M.; Dies-Suarez, P. Resolution of fatty liver disease after growth hormone replacement in a pediatric survivor of thyroid cancer. Bol. Med. Hosp. Infant. Mex. 2019, 76, 138–145.
- Takano, S.; Kanzaki, S.; Sato, M.; Kubo, T.; Seino, Y. Effect of growth hormone on fatty liver in panhypopituitarism. Arch. Dis. Child. 1997, 76, 537–538.
- Fujio, A.; Kawagishi, N.; Echizenya, T.; Tokodai, K.; Nakanishi, C.; Miyagi, S.; Sato, K.; Fujimori, K.; Ohuchi, N. Long-term survival with growth hormone replacement after liver transplantation of pediatric nonalcoholic steatohepatitis complicating acquired hypopituitarism. Tohoku J. Exp. Med. 2015, 235, 61–67.
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74.
- Rufinatscha, K.; Ress, C.; Folie, S.; Haas, S.; Salzmann, K.; Moser, P.; Dobner, J.; Weiss, G.; Iruzubieta, P.; Arias-Loste, M.T.; et al. Metabolic effects of reduced growth hormone action in fatty liver disease. Hepatol. Int. 2018, 12, 474–481.
- Guichelaar, M.M.; Charlton, M.R. Decreased muscle mass in nonalcoholic fatty liver disease: New evidence of a link between growth hormone and fatty liver disease? Hepatology 2014, 59, 1668–1670.
- Fukunaga, K.; Imachi, H.; Lyu, J.; Dong, T.; Sato, S.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Yonezaki, K.; Matsunaga, T.; et al. IGF1 suppresses cholesterol accumulation in the liver of growth hormone-deficient mice via the activation of ABCA1. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1232–E1241.
- Sanz, S.; Pucilowska, J.B.; Liu, S.; Rodriguez-Ortigosa, C.M.; Lund, P.K.; Brenner, D.A.; Fuller, C.R.; Simmons, J.G.; Pardo, A.; Martinez-Chantar, M.L.; et al. Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut 2005, 54, 134–141.
- Sumida, Y.; Yonei, Y.; Tanaka, S.; Mori, K.; Kanemasa, K.; Imai, S.; Taketani, H.; Hara, T.; Seko, Y.; Ishiba, H.; et al. Lower levels of insulin-like growth factor-1 standard deviation score are associated with histological severity of non-alcoholic fatty liver disease. Hepatol. Res. 2015, 45, 771–781.
- Ukropec, J.; Penesova, A.; Skopkova, M.; Pura, M.; Vlcek, M.; Radikova, Z.; Imrich, R.; Ukropcova, B.; Tajtakova, M.; Koska, J.; et al. Adipokine protein expression pattern in growth hormone deficiency predisposes to the increased fat cell size and the whole body metabolic derangements. J. Clin. Endocrinol. Metab. 2008, 93, 2255–2262.
- Arturi, F.; Succurro, E.; Procopio, C.; Pedace, E.; Mannino, G.C.; Lugara, M.; Procopio, T.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J. Clin. Endocrinol. Metab. 2011, 96, E1640–E1644.
- Pan, C.S.; Weiss, J.J.; Fourman, L.T.; Buckless, C.; Branch, K.L.; Lee, H.; Torriani, M.; Misra, M.; Stanley, T.L. Effect of recombinant human growth hormone on liver fat content in young adults with nonalcoholic fatty liver disease. Clin. Endocrinol. (Oxf.) 2021, 94, 183–192.
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605.
- Cabrera, D.; Cabello-Verrugio, C.; Solis, N.; San Martin, D.; Cofre, C.; Pizarro, M.; Arab, J.P.; Abrigo, J.; Campos, F.; Irigoyen, B.; et al. Somatotropic Axis Dysfunction in Non-Alcoholic Fatty Liver Disease: Beneficial Hepatic and Systemic Effects of Hormone Supplementation. Int. J. Mol. Sci. 2018, 19, 1339.
- Petrossians, P.; Daly, A.F.; Natchev, E.; Maione, L.; Blijdorp, K.; Sahnoun-Fathallah, M.; Auriemma, R.; Diallo, A.M.; Hulting, A.L.; Ferone, D.; et al. Acromegaly at diagnosis in 3173 patients from the Liege Acromegaly Survey (LAS) Database. Endocr. Relat. Cancer 2017, 24, 505–518.
- Moller, N.; Jorgensen, J.O. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177.
- Winhofer, Y.; Wolf, P.; Krssak, M.; Wolfsberger, S.; Tura, A.; Pacini, G.; Gessl, A.; Raber, W.; Kukurova, I.J.; Kautzky-Willer, A.; et al. No evidence of ectopic lipid accumulation in the pathophysiology of the acromegalic cardiomyopathy. J. Clin. Endocrinol. Metab. 2014, 99, 4299–4306.
- Koutsou-Tassopoulou, A.; Papapostoli-Sklavounou, I.; Krawczyk, M.; Friesenhahn-Ochs, B.; Weber, S.N.; Lammert, F.; Stokes, C.S. Hepatic steatosis in patients with acromegaly. Endocrinol. Diabetes Metab. 2019, 2, e00090.
- Drake, W.M.; Rowles, S.V.; Roberts, M.E.; Fode, F.K.; Besser, G.M.; Monson, J.P.; Trainer, P.J. Insulin sensitivity and glucose tolerance improve in patients with acromegaly converted from depot octreotide to pegvisomant. Eur. J. Endocrinol. 2003, 149, 521–527.
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720.
- Neggers, S.J.; Kopchick, J.J.; Jorgensen, J.O.; van der Lely, A.J. Hypothesis: Extra-hepatic acromegaly: A new paradigm? Eur. J. Endocrinol. 2011, 164, 11–16.
- Rubeck, K.Z.; Madsen, M.; Andreasen, C.M.; Fisker, S.; Frystyk, J.; Jorgensen, J.O. Conventional and novel biomarkers of treatment outcome in patients with acromegaly: Discordant results after somatostatin analog treatment compared with surgery. Eur. J. Endocrinol. 2010, 163, 717–726.
- Flyvbjerg, A.; Bennett, W.F.; Rasch, R.; Kopchick, J.J.; Scarlett, J.A. Inhibitory effect of a growth hormone receptor antagonist (G120K-PEG) on renal enlargement, glomerular hypertrophy, and urinary albumin excretion in experimental diabetes in mice. Diabetes 1999, 48, 377–382.
- Moller, L.; Norrelund, H.; Jessen, N.; Flyvbjerg, A.; Pedersen, S.B.; Gaylinn, B.D.; Liu, J.; Thorner, M.O.; Moller, N.; Lunde Jorgensen, J.O. Impact of growth hormone receptor blockade on substrate metabolism during fasting in healthy subjects. J. Clin. Endocrinol. Metab. 2009, 94, 4524–4532.
- Droste, M.; Domberg, J.; Buchfelder, M.; Mann, K.; Schwanke, A.; Stalla, G.; Strasburger, C.J. Therapy of acromegalic patients exacerbated by concomitant type 2 diabetes requires higher pegvisomant doses to normalise IGF1 levels. Eur. J. Endocrinol. 2014, 171, 59–68.
- Szendroedi, J.; Zwettler, E.; Schmid, A.I.; Chmelik, M.; Pacini, G.; Kacerovsky, G.; Smekal, G.; Nowotny, P.; Wagner, O.; Schnack, C.; et al. Reduced basal ATP synthetic flux of skeletal muscle in patients with previous acromegaly. PLoS ONE 2008, 3, e3958.
- Ciresi, A.; Guarnotta, V.; Campo, D.; Giordano, C. Hepatic Steatosis Index in Acromegaly: Correlation with Insulin Resistance Regardless of the Disease Control. Int. J. Endocrinol. 2018, 2018, 5421961.
- Bernard, V.; Young, J.; Binart, N. Prolactin—A pleiotropic factor in health and disease. Nat. Rev. Endocrinol. 2019, 15, 356–365.
- Macotela, Y.; Triebel, J.; Clapp, C. Time for a New Perspective on Prolactin in Metabolism. Trends Endocrinol. Metab. 2020, 31, 276–286.
- Freemark, M.; Avril, I.; Fleenor, D.; Driscoll, P.; Petro, A.; Opara, E.; Kendall, W.; Oden, J.; Bridges, S.; Binart, N.; et al. Targeted deletion of the PRL receptor: Effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002, 143, 1378–1385.
- Ruiz-Herrera, X.; de Los Rios, E.A.; Diaz, J.M.; Lerma-Alvarado, R.M.; Martinez de la Escalera, L.; Lopez-Barrera, F.; Lemini, M.; Arnold, E.; Martinez de la Escalera, G.; Clapp, C.; et al. Prolactin Promotes Adipose Tissue Fitness and Insulin Sensitivity in Obese Males. Endocrinology 2017, 158, 56–68.
- Serri, O.; Li, L.; Mamputu, J.C.; Beauchamp, M.C.; Maingrette, F.; Renier, G. The influences of hyperprolactinemia and obesity on cardiovascular risk markers: Effects of cabergoline therapy. Clin. Endocrinol. (Oxf.) 2006, 64, 366–370.
- Berinder, K.; Nystrom, T.; Hoybye, C.; Hall, K.; Hulting, A.L. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary 2011, 14, 199–207.
- dos Santos Silva, C.M.; Barbosa, F.R.; Lima, G.A.; Warszawski, L.; Fontes, R.; Domingues, R.C.; Gadelha, M.R. BMI and metabolic profile in patients with prolactinoma before and after treatment with dopamine agonists. Obesity 2011, 19, 800–805.
- Nagano, M.; Kelly, P.A. Tissue distribution and regulation of rat prolactin receptor gene expression. Quantitative analysis by polymerase chain reaction. J. Biol. Chem. 1994, 269, 13337–13345.
- Zhang, P.; Ge, Z.; Wang, H.; Feng, W.; Sun, X.; Chu, X.; Jiang, C.; Wang, Y.; Zhu, D.; Bi, Y. Prolactin improves hepatic steatosis via CD36 pathway. J. Hepatol. 2018, 68, 1247–1255.
- Shao, S.; Yao, Z.; Lu, J.; Song, Y.; He, Z.; Yu, C.; Zhou, X.; Zhao, L.; Zhao, J.; Gao, L. Ablation of prolactin receptor increases hepatic triglyceride accumulation. Biochem. Biophys. Res. Commun. 2018, 498, 693–699.
- Yu, J.; Xiao, F.; Zhang, Q.; Liu, B.; Guo, Y.; Lv, Z.; Xia, T.; Chen, S.; Li, K.; Du, Y.; et al. PRLR regulates hepatic insulin sensitivity in mice via STAT5. Diabetes 2013, 62, 3103–3113.
- Luque, G.M.; Lopez-Vicchi, F.; Ornstein, A.M.; Brie, B.; De Winne, C.; Fiore, E.; Perez-Millan, M.I.; Mazzolini, G.; Rubinstein, M.; Becu-Villalobos, D. Chronic hyperprolactinemia evoked by disruption of lactotrope dopamine D2 receptors impacts on liver and adipocyte genes related to glucose and insulin balance. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E974–E988.
- Park, S.; Kim, D.S.; Daily, J.W.; Kim, S.H. Serum prolactin concentrations determine whether they improve or impair beta-cell function and insulin sensitivity in diabetic rats. Diabetes Metab Res Rev 2011, 27, 564–574.
- Christ-Crain, M. Vasopressin and Copeptin in health and disease. Rev. Endocr. Metab. Disord. 2019, 20, 283–294.
- Nakamura, K.; Velho, G.; Bouby, N. Vasopressin and metabolic disorders: Translation from experimental models to clinical use. J. Intern. Med. 2017, 282, 298–309.
- Enhorning, S.; Melander, O. The Vasopressin System in the Risk of Diabetes and Cardiorenal Disease, and Hydration as a Potential Lifestyle Intervention. Ann. Nutr. Metab. 2018, 72 (Suppl. 2), 21–27.
- Barchetta, I.; Enhorning, S.; Cimini, F.A.; Capoccia, D.; Chiappetta, C.; Di Cristofano, C.; Silecchia, G.; Leonetti, F.; Melander, O.; Cavallo, M.G. Elevated plasma copeptin levels identify the presence and severity of non-alcoholic fatty liver disease in obesity. BMC Med. 2019, 17, 85.
- Hems, D.A.; Whitton, P.D. Stimulation by vasopressin of glycogen breakdown and gluconeogenesis in the perfused rat liver. Biochem. J. 1973, 136, 705–709.
- Fisher, R.A.; Robertson, S.M.; Olson, M.S. Stimulation of glycogenolysis and vasoconstriction in the perfused rat liver by the thromboxane A2 analogue U-46619. J. Biol. Chem. 1987, 262, 4631–4638.
- Johnson, E.C.; Bardis, C.N.; Jansen, L.T.; Adams, J.D.; Kirkland, T.W.; Kavouras, S.A. Reduced water intake deteriorates glucose regulation in patients with type 2 diabetes. Nutr. Res. 2017, 43, 25–32.
- Hiroyama, M.; Aoyagi, T.; Fujiwara, Y.; Birumachi, J.; Shigematsu, Y.; Kiwaki, K.; Tasaki, R.; Endo, F.; Tanoue, A. Hypermetabolism of fat in V1a vasopressin receptor knockout mice. Mol. Endocrinol. 2007, 21, 247–258.
- Rofe, A.M.; Williamson, D.H. Metabolic effects of vasopressin infusion in the starved rat. Reversal of ketonaemia. Biochem. J. 1983, 212, 231–239.
- Xue, B.; Moustaid, N.; Wilkison, W.O.; Zemel, M.B. The agouti gene product inhibits lipolysis in human adipocytes via a Ca2+-dependent mechanism. FASEB J. 1998, 12, 1391–1396.
- Palmer, T.N.; Caldecourt, M.A.; Watts, D.I.; Sugden, M.C. Inhibition of lipogenesis by vasopressin and angiotensin II in glycogen-depleted hepatocytes. Biosci. Rep. 1983, 3, 1063–1070.
- Taveau, C.; Chollet, C.; Waeckel, L.; Desposito, D.; Bichet, D.G.; Arthus, M.F.; Magnan, C.; Philippe, E.; Paradis, V.; Foufelle, F.; et al. Vasopressin and hydration play a major role in the development of glucose intolerance and hepatic steatosis in obese rats. Diabetologia 2015, 58, 1081–1090.
- Alshahrani, F.; Aljohani, N. Vitamin D: Deficiency, sufficiency and toxicity. Nutrients 2013, 5, 3605–3616.
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689S–1696S.
- Cimini, F.A.; Barchetta, I.; Carotti, S.; Bertoccini, L.; Baroni, M.G.; Vespasiani-Gentilucci, U.; Cavallo, M.G.; Morini, S. Relationship between adipose tissue dysfunction, vitamin D deficiency and the pathogenesis of non-alcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 3407–3417.
- Wimalawansa, S.J. Associations of vitamin D with insulin resistance, obesity, type 2 diabetes, and metabolic syndrome. J. Steroid Biochem. Mol. Biol. 2018, 175, 177–189.
- Prasad, P.; Kochhar, A. Interplay of vitamin D and metabolic syndrome: A review. Diabetes Metab. Syndr. 2016, 10, 105–112.
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Ben Tov, A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737.
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D counteracts fibrogenic TGF-beta signalling in human hepatic stellate cells both receptor-dependently and independently. Gut 2015, 64, 791–799.
- Dong, B.; Zhou, Y.; Wang, W.; Scott, J.; Kim, K.; Sun, Z.; Guo, Q.; Lu, Y.; Gonzales, N.M.; Wu, H.; et al. Vitamin D Receptor Activation in Liver Macrophages Ameliorates Hepatic Inflammation, Steatosis, and Insulin Resistance in Mice. Hepatology 2019, 71, 1559–1574.
- Bozic, M.; Guzman, C.; Benet, M.; Sanchez-Campos, S.; Garcia-Monzon, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016, 65, 748–757.
- Eliades, M.; Spyrou, E.; Agrawal, N.; Lazo, M.; Brancati, F.L.; Potter, J.J.; Koteish, A.A.; Clark, J.M.; Guallar, E.; Hernaez, R. Meta-analysis: Vitamin D and non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 38, 246–254.
- Liangpunsakul, S.; Chalasani, N. Serum vitamin D concentrations and unexplained elevation in ALT among US adults. Dig. Dis. Sci. 2011, 56, 2124–2129.
- Eliades, M.; Spyrou, E. Vitamin D: A new player in non-alcoholic fatty liver disease? World J. Gastroenterol. 2015, 21, 1718–1727.
- Targher, G.; Bertolini, L.; Scala, L.; Cigolini, M.; Zenari, L.; Falezza, G.; Arcaro, G. Associations between serum 25-hydroxyvitamin D3 concentrations and liver histology in patients with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 517–524.
- Liu, S.; Liu, Y.; Wan, B.; Zhang, H.; Wu, S.; Zhu, Z.; Lin, Y.; Wang, M.; Zhang, N.; Lin, S.; et al. Association between Vitamin D Status and Non-Alcoholic Fatty Liver Disease: A Population-Based Study. J. Nutr. Sci. Vitaminol. (Tokyo) 2019, 65, 303–308.
- Borges-Canha, M.; Neves, J.S.; Mendonça, F.; Silva, M.M.; Costa, C.; Cabral, P.M.; Guerreiro, V.; Lourenço, R.; Meira, P.; Salazar, D.; et al. The Impact of Vitamin D in Non-Alcoholic Fatty Liver Disease: A Cross-Sectional Study in Patients with Morbid Obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 487–495.
- Ha, Y.; Hwang, S.G.; Rim, K.S. The Association between Vitamin D Insufficiency and Nonalcoholic Fatty Liver Disease: A Population-Based Study. Nutrients 2017, 9, 806.
- Wang, N.; Chen, C.; Zhao, L.; Chen, Y.; Han, B.; Xia, F.; Cheng, J.; Li, Q.; Lu, Y. Vitamin D and Nonalcoholic Fatty Liver Disease: Bi-directional Mendelian Randomization Analysis. EBioMedicine 2018, 28, 187–193.
- Barchetta, I.; Del Ben, M.; Angelico, F.; Di Martino, M.; Fraioli, A.; La Torre, G.; Saulle, R.; Perri, L.; Morini, S.; Tiberti, C.; et al. No effects of oral vitamin D supplementation on non-alcoholic fatty liver disease in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled trial. BMC Med. 2016, 14, 92.
- Han, H.; Cui, M.; You, X.; Chen, M.; Piao, X.; Jin, G. A role of 1,25(OH)2D3 supplementation in rats with nonalcoholic steatohepatitis induced by choline-deficient diet. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 556–561.
- Keane, J.T.; Elangovan, H.; Stokes, R.A.; Gunton, J.E. Vitamin D and the Liver-Correlation or Cause? Nutrients 2018, 10, 496.
- Dabbaghmanesh, M.H.; Danafar, F.; Eshraghian, A.; Omrani, G.R. Vitamin D supplementation for the treatment of non-alcoholic fatty liver disease: A randomized double blind placebo controlled trial. Diabetes Metab. Syndr. 2018, 12, 513–517.
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015.
- Bjelakovic, G.; Nikolova, D.; Bjelakovic, M.; Gluud, C. Vitamin D supplementation for chronic liver diseases in adults. Cochrane Database Syst. Rev. 2017, 11, CD011564.
- Pacifico, L.; Osborn, J.F.; Bonci, E.; Pierimarchi, P.; Chiesa, C. Association between Vitamin D Levels and Nonalcoholic Fatty Liver Disease: Potential Confounding Variables. Mini. Rev. Med. Chem. 2019, 19, 310–332.
- Filip, R.; Radzki, R.P.; Bienko, M. Novel insights into the relationship between nonalcoholic fatty liver disease and osteoporosis. Clin. Interv. Aging 2018, 13, 1879–1891.
- Targher, G.; Lonardo, A.; Rossini, M. Nonalcoholic fatty liver disease and decreased bone mineral density: Is there a link? J. Endocrinol. Investig. 2015, 38, 817–825.
- Poggiogalle, E.; Donini, L.M.; Lenzi, A.; Chiesa, C.; Pacifico, L. Non-alcoholic fatty liver disease connections with fat-free tissues: A focus on bone and skeletal muscle. World J. Gastroenterol. 2017, 23, 1747–1757.
- Xia, M.F.; Lin, H.D.; Yan, H.M.; Bian, H.; Chang, X.X.; Zhang, L.S.; He, W.Y.; Gao, X. The association of liver fat content and serum alanine aminotransferase with bone mineral density in middle-aged and elderly Chinese men and postmenopausal women. J. Transl. Med. 2016, 14, 11.
- Li, M.; Xu, Y.; Xu, M.; Ma, L.; Wang, T.; Liu, Y.; Dai, M.; Chen, Y.; Lu, J.; Liu, J.; et al. Association between nonalcoholic fatty liver disease (NAFLD) and osteoporotic fracture in middle-aged and elderly Chinese. J. Clin. Endocrinol. Metab. 2012, 97, 2033–2038.
- Lee, S.H.; Yun, J.M.; Kim, S.H.; Seo, Y.G.; Min, H.; Chung, E.; Bae, Y.S.; Ryou, I.S.; Cho, B. Association between bone mineral density and nonalcoholic fatty liver disease in Korean adults. J. Endocrinol. Investig. 2016, 39, 1329–1336.
- Cui, R.; Sheng, H.; Rui, X.F.; Cheng, X.Y.; Sheng, C.J.; Wang, J.Y.; Qu, S. Low bone mineral density in chinese adults with nonalcoholic Fatty liver disease. Int. J. Endocrinol. 2013, 2013, 396545.
- Moon, S.S.; Lee, Y.S.; Kim, S.W. Association of nonalcoholic fatty liver disease with low bone mass in postmenopausal women. Endocrine 2012, 42, 423–429.
- Bhatt, S.P.; Nigam, P.; Misra, A.; Guleria, R.; Qadar Pasha, M.A. Independent associations of low 25 hydroxy vitamin D and high parathyroid hormonal levels with nonalcoholic fatty liver disease in Asian Indians residing in north India. Atherosclerosis 2013, 230, 157–163.
- Upala, S.; Jaruvongvanich, V.; Wijarnpreecha, K.; Sanguankeo, A. Nonalcoholic fatty liver disease and osteoporosis: A systematic review and meta-analysis. J. Bone Miner. Metab. 2017, 35, 685–693.
More
Information
Subjects:
Nutrition & Dietetics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
856
Revisions:
2 times
(View History)
Update Date:
15 Apr 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No