 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanni Luca Beretta | -- | 2822 | 2022-04-13 11:22:10 | | | |
| 2 | Peter Tang | -3 word(s) | 2819 | 2022-04-13 11:50:21 | | |
Video Upload Options
Necroptosis is a programmed form of necrosis characterized by mitochondrial alterations and plasma membrane permeabilization resulting in the release of cytoplasmic content into extracellular space, and leading to inflammatory reactions. Besides its critical role in viral defense mechanisms and inflammatory diseases, necroptosis plays pivotal functions in the drug response of tumors, including prostate cancer. Necroptosis is mainly governed by kinase enzymes, including RIP1, RIP3, and MLKL, and conversely to apoptosis, is a caspase-independent mechanism of cell death. Numerous compounds induce necroptosis in prostate cancer models, including (i) compounds of natural origin, (ii) synthetic and semisynthetic small molecules, and (iii) selenium and selenium-based nanoparticles.
1. Introduction
2. Apoptosis and Necroptosis: Overview on Molecular Mechanisms
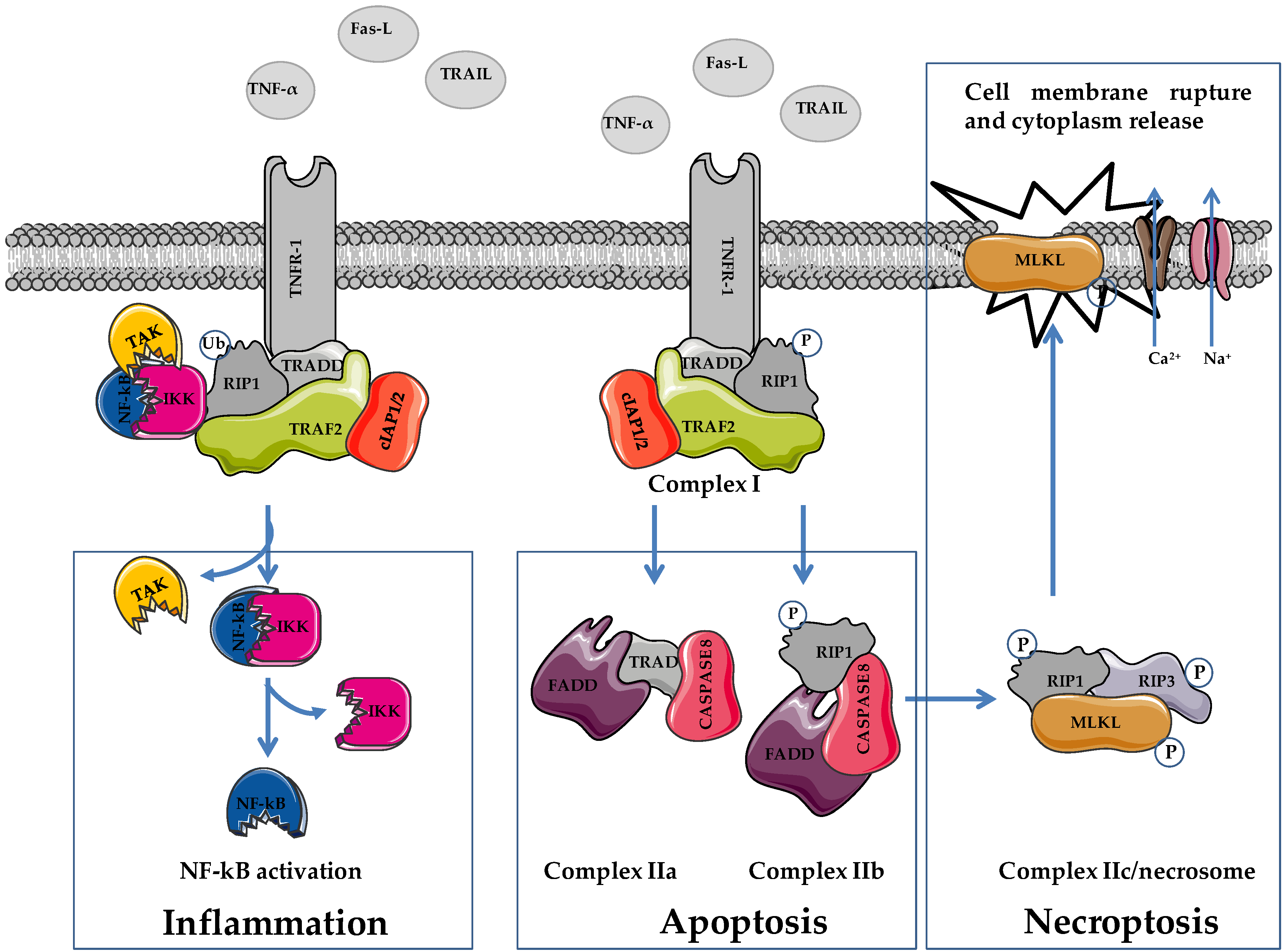
|
Characteristic |
Necroptosis |
Necrosis |
Apoptosis |
|
|---|---|---|---|---|
|
Involved proteins |
RIP3 |
+ |
/ |
/ |
|
MLKL |
+ |
/ |
/ |
|
|
Caspase 3 |
/ |
/ |
+ |
|
|
Cell properties |
Membrane perforation |
+ |
+ |
/ |
|
Membrane blebbing |
/ |
/ |
+ |
|
|
DNA fragmentation |
+ |
+ |
+ |
|
|
Cell lysis and swelling |
+ |
+ |
/ |
|
|
Inflammation |
+ |
+ |
/ |
3. Necroptosis and Necroptosis Inducers in Prostate Cancer
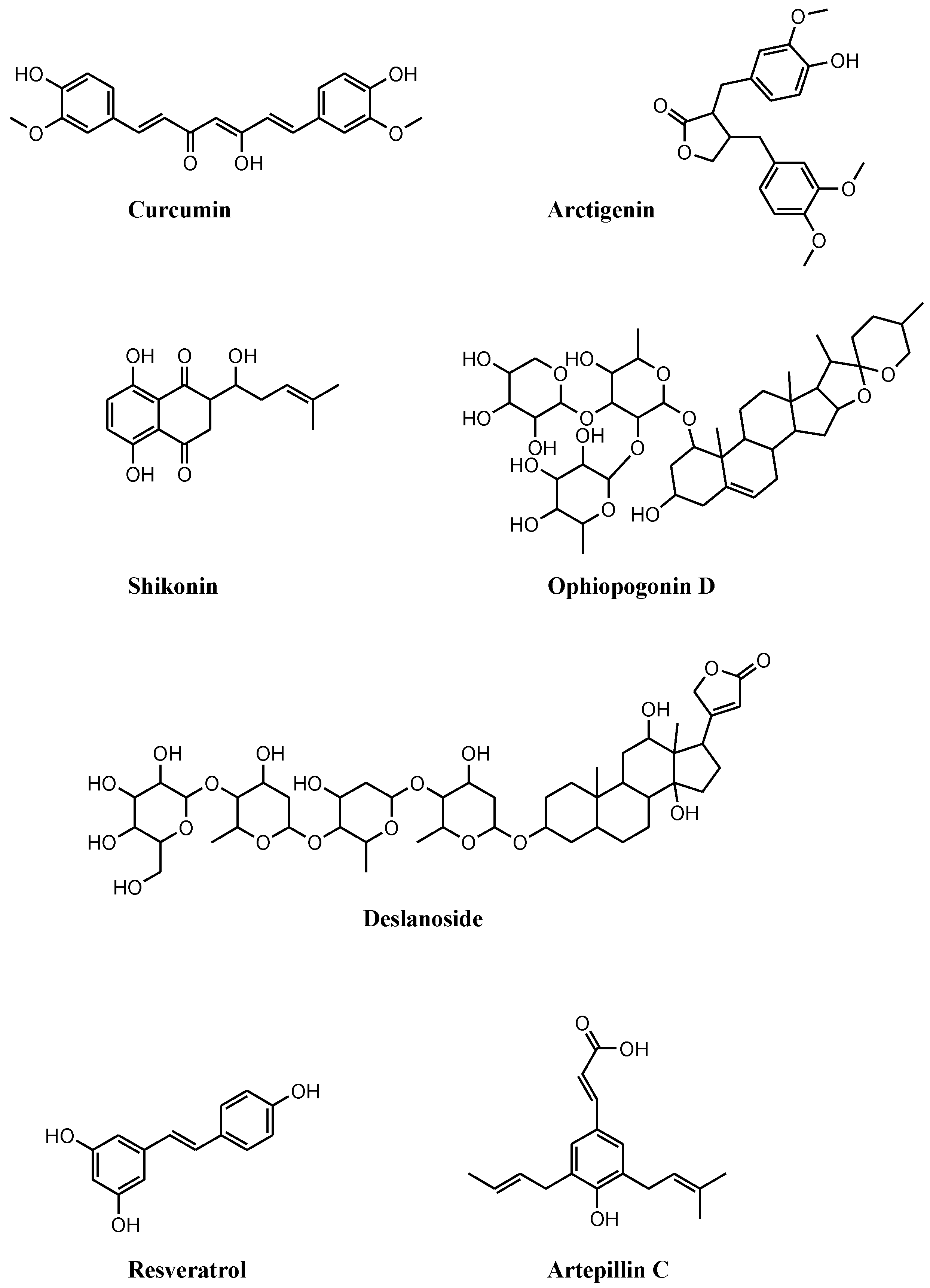
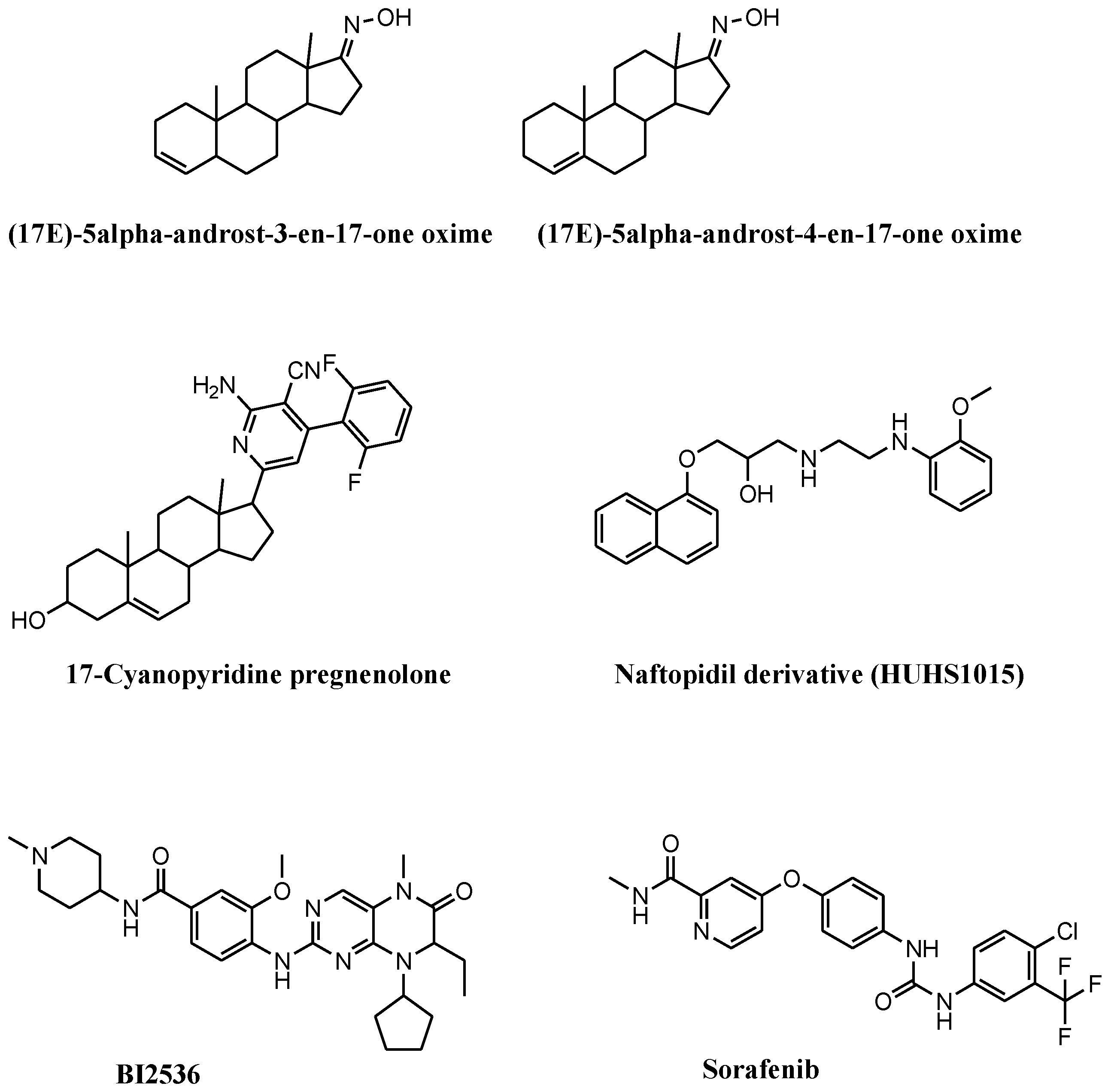
4. Ongoing Clinical Trials Containing Necroptosis Inducers in Prostate Cancer
|
Compound |
NCT Number |
Markers |
Phase |
|---|---|---|---|
|
Curcumin |
NCT03769766 |
PSA |
III |
|
NCT03211104 |
PSA |
na |
|
|
NCT02064673 |
PSA |
III |
|
|
Curcumin and piperine |
NCT04731844 |
nd |
II |
|
Curcumin and ursolic acid |
NCT04403568 |
p65, NF-kB |
I |
|
Curcumin and Vitamin D, omega 3, turmeric |
NCT03290417 |
PSA |
na |
|
Curcumin and taxotere |
NCT02095717 |
PSA |
III |
|
Curcumin and radiotherapy |
NCT01917890 |
TNF-α, NF-kB |
na |
|
NCT02724618 |
PSA |
II |
|
|
NCT03493997 |
nd |
II |
|
|
Polyphenon E |
NCT00596011 |
PSA |
II |
|
NCT00676780 |
PSA, VEGF, HGF |
II |
|
|
NCT01340599 |
PSA, Ki67, Bcl2, Cyclin D, p27, VEGF, CD31, MMP2 and 9, IGF1 |
II |
|
|
NCT00459407 |
MMP2, MMP9, IGF1 |
I |
|
|
NCT00253643 |
FASN, Ki67 |
na |
|
|
NCT04597359 |
PSA, Ki67 |
II |
|
|
BI2536 |
NCT00706498 |
PSA |
II |
|
Sorafenib |
NCT00090545 |
PSA |
II |
|
NCT00694291 |
PSA |
II |
|
|
NCT00466752 |
PSA, p-ERK, p-AKT, p-S6-kinase, caspase 3, Ki67 |
I |
|
|
NCT00093457 |
PSA |
II |
|
|
Sorafenib and leuprolide or bicalutamide |
NCT00924807 |
PSA |
I-II |
|
Sorafenib and docetaxel |
NCT00589420 |
PSA |
II |
|
NCT00619996 |
PSA, p-ERK, VEGF-R2 |
II |
|
|
Sorafenib and taxotere |
NCT00405210 |
PSA |
I |
|
Sorafenib and mitoxantrone |
NCT00452387 |
PSA |
II |
|
Sorafenib and gleevec |
NCT00424385 |
PSA |
I |
|
Selenite and docetaxel |
NCT01155791 |
PSA |
I |
|
Selenite and radiotherapy |
NCT02184533 |
PSA |
I |
References
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364.
- Choi, J.J.; Reich, C.F.; Pisetsky, D.S. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 2004, 60, 159–166.
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host. Microbe. 2018, 24, 97–108.e104.
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood J. Am. Soc. Hematol. 2016, 127, 139–148.
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64.
- Wang, Y.; Wu, N.; Jiang, N. Autophagy provides a conceptual therapeutic framework for bone metastasis from prostate cancer. Cell Death Dis. 2021, 12, 909.
- Zaffaroni, N.; Beretta, G.L. Nanoparticles for Ferroptosis Therapy in Cancer. Pharmaceutics 2021, 13, 1785.
- Zaffaroni, N.; Beretta, G.L. Ferroptosis inducers for prostate cancer therapy. Curr. Med. Chem. 2022, in press.
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy RIPs into cell death. Cell Cycle 2016, 15, 3014–3015.
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720.
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49.
- Heidaryan, F.; Bamehr, H.; Babaabasi, B.; Emamvirdizadeh, A.; Mohammadzadeh, N.; Khalili, A. The Trend of ripk1/ripk3 and mlkl Mediated Necroptosis Pathway in Patients with Different Stages of Prostate Cancer as Promising Progression Biomarkers. Clin. Lab. 2020, 66.
- Liu, W.; Jin, W.; Zhu, S.; Chen, Y.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in cancer therapy: A revisited review of apoptosis, autophagy-dependent cell death and necroptosis. Drug Discov. Today 2022, 27, 612–625.
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417.
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124.
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.G. Necroptosis and tumor progression. Trends Cancer 2022, 8, 21–27.
- Nie, Z.; Chen, M.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Zhang, S. Regulated Cell Death in Urinary Malignancies. Front. Cell Dev. Biol. 2021, 9, 789004.
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on systemic prostate cancer therapies: Management of metastatic castration resistant prostate cancer in the era of precision oncology. Eur. Urol. 2019, 75, 88–99.
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Sanchez-Niño, M.D.; Ramos, A.M.; Cabello, R.; Gonzalez-Enguita, C.; Linkermann, A.; Sanz, A.B.; Ortiz, A. Cell death-based approaches in treatment of the urinary tract-associated diseases: A fight for survival in the killing fields. Cell Death Dis. 2018, 9, 118.
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Liu, X.; Xie, X.; Ren, Y.; Shao, Z.; Zhang, N.; Li, L.; Ding, X.; Zhang, L. The role of necroptosis in disease and treatment. MedComm 2021, 2, 730–755.
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171.
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147.
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634.
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930.
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495.
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130.
- Dondelinger, Y.; Darding, M.; Bertrand, M.J.; Walczak, H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol. Life Sci. 2016, 73, 2165–2176.
- Tortola, L.; Nitsch, R.; Bertrand, M.J.M.; Kogler, M.; Redouane, Y.; Kozieradzki, I.; Uribesalgo, I.; Fennell, L.M.; Daugaard, M.; Klug, H.; et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016, 16, 3414.
- Zhang, K.X.; Firus, J.; Prieur, B.; Jia, W.; Rennie, P.S. To die or to survive, a fatal question for the destiny of prostate cancer cells after androgen deprivation therapy. Cancers 2011, 3, 1498–1512.
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106.
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545.
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407.
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 22, 971–981.
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601.
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227.
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243.
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197.
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885.
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23.
- Wang, K.J.; Wang, K.Y.; Zhang, H.Z.; Meng, X.Y.; Chen, J.F.; Wang, P.; Jiang, J.H.; Ma, Q. Up-Regulation of RIP3 Alleviates Prostate Cancer Progression by Activation of RIP3/MLKL Signaling Pathway and Induction of Necroptosis. Front. Oncol. 2020, 10, 1720.
- Lu, Z.; Wu, C.; Zhu, M.; Song, W.; Wang, H.; Wang, J.; Guo, J.; Li, N.; Liu, J.; Li, Y.; et al. Ophiopogonin D' induces RIPK1-dependent necroptosis in androgen-dependent LNCaP prostate cancer cells. Int J. Oncol. 2020, 56, 439–447.
- Fu, W.; Li, H.; Fu, H.; Zhao, S.; Shi, W.; Sun, M.; Li, Y. The SIRT3 and SIRT6 Promote Prostate Cancer Progression by Inhibiting Necroptosis-Mediated Innate Immune Response. J. Immunol. Res. 2020, 2020, 8820355.


