Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rohan Rao | -- | 2927 | 2022-04-12 21:52:16 | | | |
| 2 | Catherine Yang | Meta information modification | 2927 | 2022-04-13 03:09:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lechpammer, M.; Rao, R.; Shah, S.; Mirheydari, M.; Bhattacharya, D.; Koehler, A.; Kamdem Toukam, D.; Haworth, K.; Pomeranz Krummel, D.; Sengupta, S. Immunotherapy for Adult Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/21670 (accessed on 28 July 2026).
Lechpammer M, Rao R, Shah S, Mirheydari M, Bhattacharya D, Koehler A, et al. Immunotherapy for Adult Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/21670. Accessed July 28, 2026.
Lechpammer, Mirna, Rohan Rao, Sanjit Shah, Mona Mirheydari, Debanjan Bhattacharya, Abigail Koehler, Donatien Kamdem Toukam, Kevin Haworth, Daniel Pomeranz Krummel, Soma Sengupta. "Immunotherapy for Adult Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/21670 (accessed July 28, 2026).
Lechpammer, M., Rao, R., Shah, S., Mirheydari, M., Bhattacharya, D., Koehler, A., Kamdem Toukam, D., Haworth, K., Pomeranz Krummel, D., & Sengupta, S. (2022, April 12). Immunotherapy for Adult Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/21670
Lechpammer, Mirna, et al. "Immunotherapy for Adult Glioblastoma." Encyclopedia. Web. 12 April, 2022.
Copy Citation
Glioblastoma, or glioblastoma multiforme (GBM, WHO Grade IV), is a highly aggressive adult glioma. Despite extensive efforts to improve treatment, the current standard-of-care (SOC) regimen, which consists of maximal resection, radiotherapy, and temozolomide (TMZ), achieves only a 12–15 month survival.
gliomas
brain tumors
immunotherapy
1. Introduction
Glioblastoma is one of the most common primary malignant adult brain tumors, typified by its aggressiveness. The current standard-of-care treatment includes maximal resection and radiotherapy, followed by adjuvant chemotherapy with the DNA alkylator temozolomide [1]. The median overall survival (MOS) following GBM diagnosis is 12–15 months [1]. A multitude of factors complicates the treatment of GBM including (1) the heterogeneous nature of the tumors, both within a patient and between patients; and (2) the highly impermeable blood–brain barrier (BBB), which limits the effective delivery of many standard therapeutics.
A recent promising advancement has been an immunotherapeutic approach, which may involve either antagonizing the tumor′s inherent immune-suppressive properties or, conversely, inducing a glioma-specific immune response using either exogenous or endogenous agents. Immunotherapy has recently been popularized for its impressive outcomes in hematogenous malignancies [2]. However, for immunotherapy to be successful in solid tumors such as GBM, it must overcome tumor heterogeneity and the physical barriers imposed by the BBB and the tumor microenvironment (TME).
2. Current Immunotherapy Options and Developments
The human immune system is a complex regulatory environment that must constantly be able to distinguish between “self” and foreign matter. The immune system can be split into “innate” and “adaptive” immunity. Innate immunity does not improve with repeated encounters and consists of phagocytic cells (neutrophils, monocytes) and pro-inflammatory cells (eosinophils, basophils, and mast cells) [3]. Adaptive immunity learns and improves upon repeated exposure to pathogens. The main players in adaptive immunity are B and T lymphocytes, which produce antigen-specific immunoglobulins and induce foreign cell lysis [3]. Following activation, part of the immune system′s natural response is to return the hyperactive immune response to basal levels. Cells such as regulatory T-cells (Tregs) release anti-inflammatory cytokines leading to a diminished immune response [4]. Similarly, cell–cell signaling via inhibitory immunoreceptors such as PD-1, CTLA-4, LAG3, TIM3, TIGIT, and BTLA can attenuate an upregulated immune response [5]. The most promising immunotherapy approaches to treating glioblastoma are immune checkpoint inhibition [6][7], T-cell transfer therapy [8], vaccination [9], and oncolytic virus therapy (OVT). These methods harness the immune system to recognize and focally target tumor cells.
2.1. Immune Checkpoint Inhibitors
Immune checkpoint inhibitors (ICIs) avert the inactivation of CD8+ T-cells by preventing checkpoint receptors from binding with their ligands (Figure 1A). The critical immune checkpoint targets for ICIs in glioma include programmed cell death protein-1 (PD-1), programmed cell death ligand-1 (PD-L1), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). In the canonical pathway, when the PD-L1 ligand on the target cells interacts with the PD-1 receptor on the T-cells, intracellular tyrosine residues on the PD-1 cytoplasmic region lead to the recruitment of Src homology 2 domain-containing protein tyrosine phosphatase-2 (SHP-2) [10]. This causes spleen tyrosine kinase (Syk) and phospholipid inositol-3-kinase (PI3K) to be phosphorylated, resulting in T-cell exhaustion and a suppressed immune response [10][11]. Glioblastoma cells can co-opt this machinery by overexpressing PD-L1, thereby evading the immune response [12]. Through a similar mechanism, glioblastoma cells also can upregulate CTLA-4, which promotes T-cell anergy through blockade of the B7/CD28 co-stimulatory signal [13].
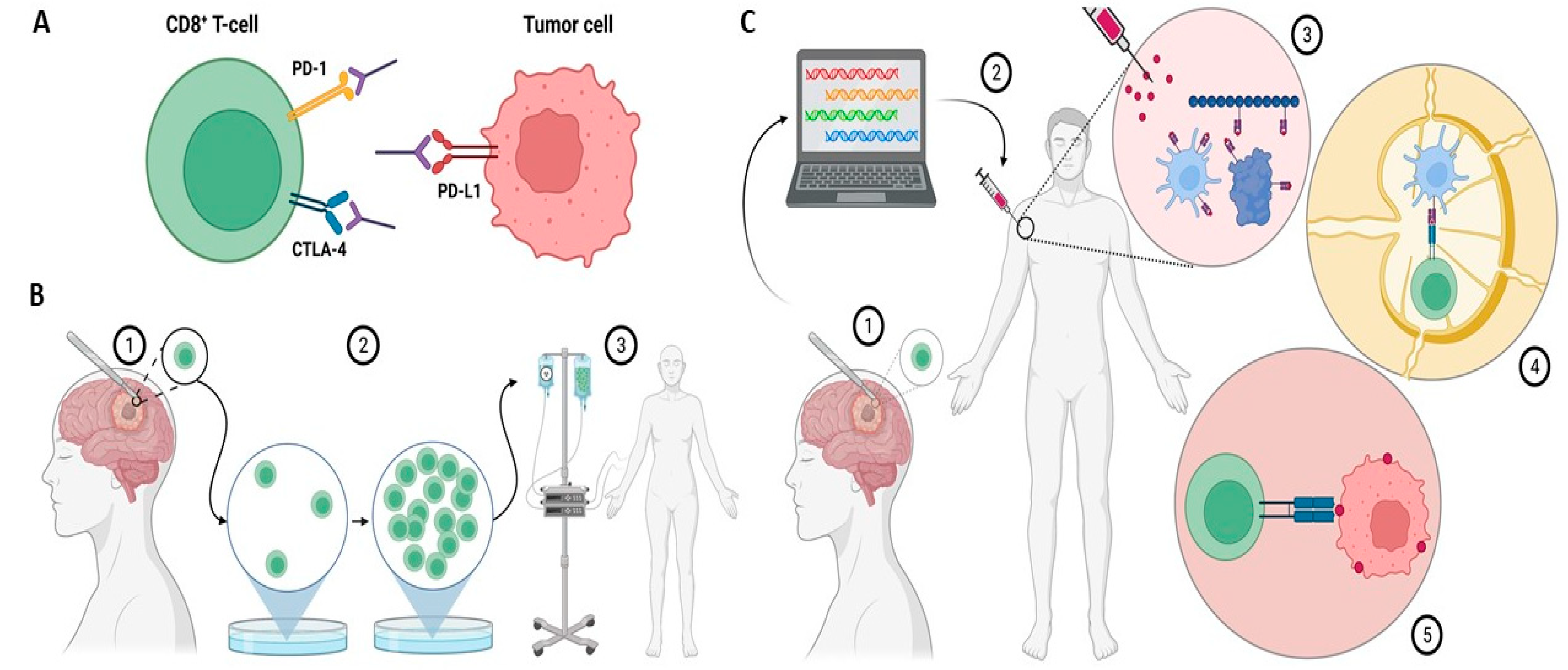
Figure 1. (A) Immune checkpoint inhibitors bind to and inhibit immunosuppressive molecules on either T-cells or tumor cells. This dampens tumor cells′ ability to evade the immune system. (B) (1) In tumor-infiltrating lymphocyte (TIL) therapy, T-cells from the tumor microenvironment are isolated following surgical resection. (2) Isolated T-cells are clonally expanded by using IL-2 stimulation. (3) Expanded T-cells are reintroduced to the patient. (C) (1) In the vaccine approach, a resected tumor biopsy is taken from the patient and sequenced to identify neoantigens. (2) Neoantigens are then delivered via a vaccine. (3) At the site of injection, neoantigens stimulate antigen-presenting cells (APCs). (4) In the lymph node, APCs present T-cells with neoantigens. (5) Activated T-cells attack cancer cells. Created with BioRender.com.
2.2. T-Cell Transfer Therapies
T-cell transfer therapy or adoptive T-cell therapy is a type of immunotherapy that encompasses two main approaches: tumor-infiltrating lymphocyte (TIL) therapy and chimeric antigen receptor (CAR) T-cell therapy (Figure 1B).
In TIL therapy, T-lymphocytes invading the TME are collected via routine biopsy or surgery, isolated using fluorescence-activated cell sorting (FACS), and then selectively expanded using IL-2 stimulation [14][15][16]. The logic behind this approach is that T-cells found in or near the tumor already have a “proven track record” for identifying cancerous cells, but there are too few of them to overcome immunosuppression. Moreover, TIL therapy significantly reduces off-target effects due to their inherent specificity to the tumor [16]. Mathewson et al. performed single-cell transcriptome sequencing in a group of patients with isocitrate dehydrogenase (IDH)-mutant and IDH-wildtype glioblastoma [17]. They described the potential effectors of anti-tumor immunity in a population of cytotoxic TILs expressing several natural killer (NK) cell genes, including the CD161-encoding gene KLRB1. The inactivation of KLRB1 or antibody-mediated CD161 blockade resulted in increased T-cell cytotoxicity against tumor cells in vitro and an enhanced response in vivo.
CAR T-cell therapy introduces synthetic T-cell receptors into T-cells, which confer the ability to recognize tumor-specific surface antigens and initiate an MHC-independent immune response [18][19][20]. CAR T-cell therapy has had great efficacy in hematogenous malignancies but has been difficult to implement in solid tumors due to the immunosuppressive environment of the TME [2][19]. Moreover, solid tumors lack highly specific surface antigens, which can lead to numerous off-target effects when using CAR T-cell therapy. Two small Phase I trials tested CAR T-cell therapy in EGFRvIII-positive recurrent glioblastoma. Although EGFRvIII-targeted CAR T-cells found their way from peripheral blood to the tumor, no meaningful response was detected [21][22]. This lack of response to anti-EGFRvIII CAR T-cells may be attributable to the significant intra- and inter-tumoral heterogeneity of EGFRvIII expression in glioblastoma as well as to adaptive changes in the local TME, which include changes in antigen expression over time. For instance, following treatment, EGFRvIII was lost in a group of patients.
2.3. Vaccination
Tumor vaccines elicit an immune response against one or several tumor antigens (Figure 1C). Vaccines usually consist of peptides or proteins, but may also constitute antigen-laden dendritic cells. Immunostimulants such as poly ICLC are often co-administered with tumor vaccines to enhance adaptive immunity.
In a single-arm, multicenter, open-label Phase I trial performed in patients with newly diagnosed Grade 3 and 4 IDH1-mutant astrocytoma, an IDH1-specific peptide vaccine induced an immune response in 30 out of 32 (93.3%) patients [23]. The 3-year progression-free and overall survival rates were 63% and 84%, respectively. The 2-year progression-free rate among patients with an immune response was 82%, while the two patients without an immune response had tumor progression within 2 years of diagnosis.
Another vaccine approach involves a vaccination against survivin, an antiapoptotic protein expressed by many tumor types [24][25][26]. Survivin expression in GBM has been associated with increased recurrence, chemotherapy resistance, and poor overall prognosis [24][25][26][27][28]. The SurVaxM vaccine contains a synthetic long peptide mimic that spans the human survivin protein sequence; it expresses MHC Class I epitopes and stimulates the MHC Class II-restricted T-cell responses required for cytotoxic CD8+ T-cell activity against tumors [24]. A Phase I trial of SurVaxM against recurrent GBM demonstrated no serious adverse events and prolonged overall survival following vaccination (86.6 weeks) compared with historical overall survival (30 weeks) [24]. A subsequent study identified that glioma patients routinely expressed elevated serum levels of CD9+/GFAP+/SVN+ exosomes, associated with tumor progression, compared with healthy controls [29]. Patients treated with antisurvivin therapy showed decreased levels of these exosomes. Monitoring of CD9+/GFAP+/SVN+ exosomes may be a promising adjunct to the use of MRI in disease surveillance. Current trials are underway to evaluate SurVaxM’s efficacy in newly diagnosed GBM [30]. However, identifying plausible new vaccine targets for GBM remains difficult due to the heterogeneity of GBM tumors.
2.4. Oncolytic Virus Therapy
In recent years, the use of OVT has shown promise in the treatment of GBMs. OVT utilizes intratumoral delivery of viral vectors to either deliver oncolytic gene therapy into the TME or to cause direct cytotoxicity through viral infection and replication [31][32]. OVT also has pro-immunogenic effects due to the induction of immunogenic cell death (ICD) in infected tumor cells. In ICD, the destruction of tumor cells by OVT leads to the release of antigenic molecules into the TME which both recruits and activates local dendritic cells, with the subsequent stimulation of specific T-cells [32].
The earliest trials of oncolytic therapy in GBM used murine fibroblasts to deliver the replication-defective herpes simplex virus 1 (HSV1) thymidine kinase (tk) gene to GBMs, which conferred increased chemosensitivity to antiviral agents such as acyclovir, ganciclovir, and valganciclovir [31][33]. However, this trial failed to show prolonged survival in the OVT group, which was hypothesized to be the result of low gene transduction rates due to the nonmigratory nature of murine fibroblasts [33]. More recently, a genetically engineered replication selective HSV1 virus, G207, has shown safety and efficacy in clinical trials. G207 contains a deletion of the diploid γ134.5 neurovirulence gene and has viral ribonucleotide reductase (UL 39) disabled by the insertion of Escherichia coli lacZ. This allows for conditional replication in tumor cells while preventing the infection of normal cells [34]. A Phase I trial showed a median survival of 15.9 months in 13 GBM patients treated with intratumoral G207, with no evidence of HSV encephalitis [35][36]. A separate Phase I trial demonstrated the safety of G207 administration in conjunction with radiotherapy [35], while a more recent trial showed its safety in the treatment of pediatric high-grade gliomas [37]. HSV-vector mediated delivery of gene therapy offers significant promise in the treatment of GBM, and a current Phase I trial is investigating the use of a new drug, rQnestin34.5v.2, after a preclinical study suggested its low toxicity to humans [38][39].
Another development in OVT was the use of intratumoral injection of aglatimagene besadenovec (GliatakTM), a replication-defective adenovirus vector-mediated delivery of HSV1-tk (AdV-tk), in conjunction with subsequent valaciclovir therapy. Phase I trials of Gliatak conducted by Chiocca and colleagues demonstrated the safety of the therapy and an impressive radiographic response [40], while the Phase II trial showed a statistically significant improvement in the MOS of GBM patients treated with Gliatak after gross total resection (GTR) compared with patients treated with the standard of care after gross total resection (25.1 months vs. 16.3 months, respectively) [41]. Importantly, the survival benefit was even further improved at 2 and 3 years compared with the standard of care treatment, but no difference was noted if the resection was subtotal [41]. However, another Phase III clinical trial named the Aspect trial, which utilized AdV-tk, showed no significant improvement in overall survival when patients were treated with intratumoral injections of AdV-tk compared with the standard of care treatment group [42]. It should be noted that the ASPECT trial had uneven use of temozolomide, and radiotherapy was not administered concomitantly with the gene therapy [42]. Yet another Phase I trial evaluated the use of a human interferon-β-expressing adenovirus vector (Ad.hIFN-β). Intratumoral injection of Ad.hIFN-β was associated with a dose-related induction of apoptosis within tumors, but several patients experienced adverse effects and one patient experienced two serious dose-related adverse effects [43]. Ultimately, further investigation into adenovirus vectors is required.
The use of a live attenuated form of poliovirus has recently been studied as well. A Phase II clinical trial demonstrated that PVSRIPO, a live attenuated poliovirus Type 1 vaccine with its cognate internal ribosome entry site replaced by that of human rhinovirus Type 2 conferred an overall survival benefit [44]. Specifically, this randomized controlled trial (RCT) showed that the group treated with PVSRIPO had an overall survival rate of 21% at both 24 and 36 months, compared with 14% and 4% in the control group, respectively [44]. The foreign ribosomal entry site on PVSRIPO causes neuronal incompetence and ablates neurovirulence [45]. The effects of PVSRIPO are mediated by CD155, a Type 1 transmembrane glycoprotein receptor that is more commonly known as the poliovirus receptor [44][46][47][48]. CD155 is almost ubiquitously upregulated in solid tumors, including GBM, and it regulates natural killer (NK) cells and is part of the Ig-superfamily adhesion family response for cell motility and invasiveness [46][48][49]. When the PV capsid binds to CD155, the capsid protein is extruded and ultimately initiates the transfer of the viral RNA genome to the cytoplasm, then subsequently allows for the translation of the RNA and mediates the viral oncolytic effects [50]. Additional Phase II studies for PVSRIPO in conjunction with additional drugs are underway, with Phase III studies likely to commence in the foreseeable future.
Translating the success of early Phase I and Phase II trials to widespread clinical use has been challenging. Phase I and Phase II trials of the drug Toca 511 (Vocimagene amiretrorepvec), a γ retroviral replicating vector encoding a transgene for an optimized yeast cytosine deaminase, demonstrated both early safety and efficacy, with prolonged overall survival and complete responses in recurrent high-grade glioma and GBM compared with accepted survival rates in the literature [51]. However, in the Phase III arm of the clinical trial, the overall survival for patients treated with Toca 511 was 11 months compared with 12 months in the patient group receiving the standard-of-care treatment, with no significant difference between the two groups [52]. Toca 511′s Phase III failure underscores how challenging the introduction of new GBM therapies into the market has been. Several obstacles underlie these challenges in translating OVT into widespread clinical use. Pre-existing antibodies and the circulating complement in the peripheral vasculature may neutralize OVT particles before they are successfully delivered into the TME [53]. Moreover, uptake into nontarget organs (e.g., the liver) is a common barrier to efficient delivery [53]. As with the other therapeutic modalities, the BBB is a major obstacle to the effective delivery of any exogenous therapeutics.
3. Strategies to Enhance Immunotherapy’s Effectiveness
Physical modalities, such as noninvasive microbubble-enhanced focused ultrasound (MB-FUS) (Figure 2), can safely and transiently alter the permeability of the BBB/BTB without directly causing changes in the tumor cells. This technology has been demonstrated preclinically in numerous species, including nonhuman primates [54][55][56][57][58][59][60][61][62][63][64], and in multiple successful Phase I and IIa clinical trials executed by several different groups [65][66][67][68][69][70][71][72][73][74]. Ultrasound-mediated BBB disruption has been observed in normal brains [54][75], brains affected by neurodegenerative diseases (e.g., Parkinson′s disease and Alzheimer′s disease) [74], and brains with tumors [65][66][76]. Together, these studies have demonstrated the robustness of the technique. The temporary increase in permeability lasts between a few hours and several days, and depends on the type and dose of the microbubbles used and the ultrasound parameters [77][78][79][80][81][82][83]. The increased permeability occurs both through the opening of the tight junctions of the endothelium and through increased transcytosis [84]. These effects are nucleated by the gentle volumetric oscillation of the microbubbles when they are exposed to low-amplitude ultrasound, with the ultrasound amplitude being within the range used for diagnostic ultrasound imaging. Care must be taken to identify the appropriate ultrasound amplitude. If the amplitudes are too low, the barrier will not be disrupted, and for amplitudes that are too high, petechial hemorrhage may occur [85]. The emissions from the oscillating microbubbles can be used to identify the appropriate amplitudes in real time, providing patient- and treatment-specific guidance and control [86][87][88][89][90][91]. Phase I clinical trials have demonstrated the safety of this technology. Oscillation of the microbubbles not only increases the permeability of the blood–brain/tumor barrier but can also establish a convective flow that enhances the delivery of chemotherapeutics [92][93][94][95].
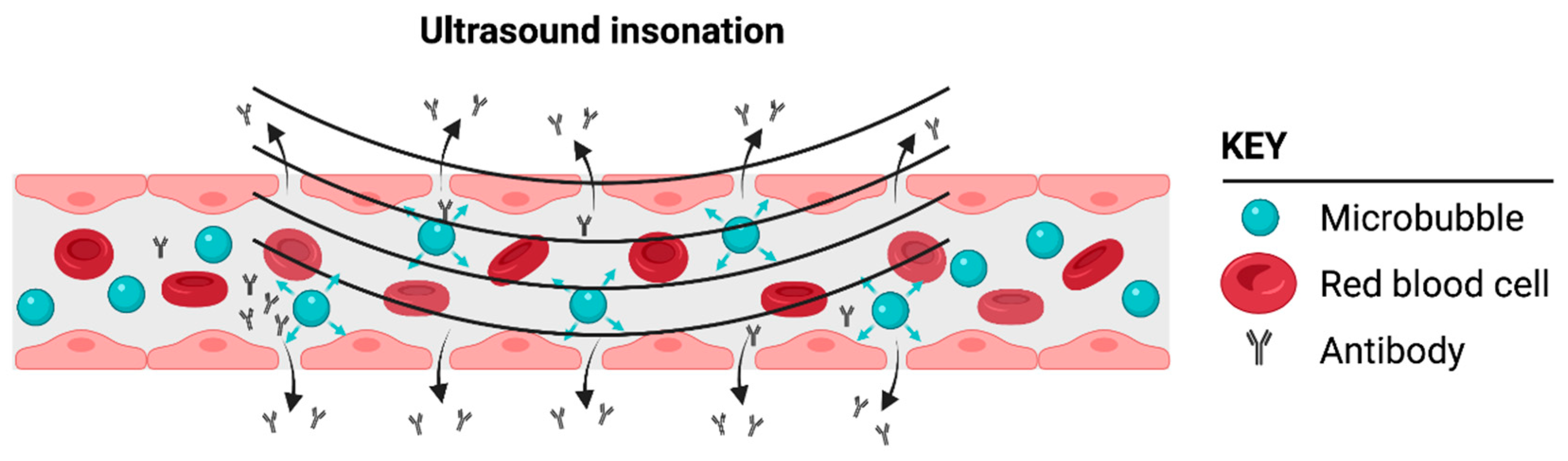
Figure 2. Cartoon illustrating how microbubbles can induce a focal disruption or opening of the blood–brain barrier (BBB), thus enabling the delivery of a biologic such as a monoclonal antibody. Microbubbles flow through the normal vasculature or vasculature supplying the glioblastoma tumor microenvironment (TME). Only the microbubbles in the vasculature exposed to ultrasound insonation enable BBB/BTB disruption following ultrasound insonation. Created with BioRender.com.
Delivery of a wide range of potential therapeutics has been demonstrated in preclinical models, including chemotherapeutics [72][96][97], adenoviruses [98][99], antibodies [100][101], nanoparticles (NPs) [94][102][103][104], and whole cells [105][106]. Guo et al. demonstrated that NPs as large as 50 nm can achieve significant extravasation into the TME with the application of focused ultrasound [94]. NPs have a wide variety of formulations. Guo et al. used them as a lipid-based encapsulation method to protect therapeutic payloads from degradation as they traversed the vasculature to the TME. Their study also demonstrated that focused ultrasound delivery of RNA-loaded NPs significantly downregulated the expression of an oncogenic mRNA [104][107]. NPs have a use in immunotherapy, as they can be combined with anti-PD-L1 antibodies to focally target drug delivery to the TME [108][109][110]. Similarly, groups have used NPs to deliver CAR-T-cells in a mouse model of glioma [109][111]. NPs could also have a use in the delivery of vaccines or OVT, given the previously discussed barriers to the effective delivery of these therapies. Ultrasound-mediated delivery to specifically induce immune modulation and therapy has been previously described [112]. Approaches include the passage of IL-12 [113], immune checkpoint inhibitors [55][67][114][115][116][117], and natural killer cells [118]. In addition to transient disruption facilitating the diffusion of therapeutics into the brain, disruption of the BBB can also enable the release of tumor biomarkers, which can assist in assessing the treatment response [119].
While there has been a significant and deserved emphasis on focused ultrasound to transiently permeabilize the blood–brain barrier, ablative ultrasound therapies can also enhance immune checkpoint inhibition [120]. Thermal ablative ultrasound therapy uses high-intensity focused ultrasound to increase the local temperature to 60 °C or higher to induce coagulative necrosis. It has been safely used in the brain to ablate neuronal tracks underlying the pathogenesis of essential tremor [121][122][123] and also to treat chronic neuropathic pain [124]. Preclinical evidence has indicated that ultrasound thermal ablation may work adjunctively with immune checkpoint inhibitors [125][126]. Furthermore, mechanically ablative ultrasound therapy (histotripsy) can also potentially enhance immune checkpoint inhibitors by stimulating nonimmunogenic “cold” tumors into becoming “hot” immunogenic tumors [127][128]. Common obstacles to this treatment approach are the interference of uniform ultrasound wave propagation through bone and gas, and organ movement during treatment, leading to collateral tissue damage [129].
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-Long Leukaemia Remissions with Persistence of CD4+ CAR T Cells. Nature 2022, 602, 503–509.
- Delves, P.J.; Roitt, I.M. The Immune System. N. Engl. J. Med. 2000, 343, 37–49.
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An Essential Role for Scurfin in CD4+CD25+ T Regulatory Cells. Nat. Immunol. 2003, 4, 337–342.
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669.
- Brahm, C.G.; van Linde, M.E.; Enting, R.H.; Schuur, M.; Otten, R.H.J.; Heymans, M.W.; Verheul, H.M.W.; Walenkamp, A.M.E. The Current Status of Immune Checkpoint Inhibitors in Neuro-Oncology: A Systematic Review. Cancers 2020, 12, 586.
- Majd, N.; Dasgupta, P.; de Groot, J. Immunotherapy for Neuro-Oncology. Adv. Exp. Med. Biol. 2020, 1244, 183–203.
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064.
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239.
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in Cancer Immunotherapy: Clinical Implications and Future Considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do They Have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296.
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 Correlates with Immune and Clinical Characteristics of Glioma. Cancer Cell Int. 2020, 20, 7.
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991.
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680.
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of Tumor-Infiltrating Lymphocyte Treatment in Solid Tumors. BMC Med. 2021, 19, 140.
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298.
- Brown, C.E.; Mackall, C.L. CAR T Cell Therapy: Inroads to Response and Resistance. Nat. Rev. Immunol. 2019, 19, 73–74.
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486.
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355.
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135.
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984.
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature 2021, 592, 463–468.
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical Study of a Survivin Long Peptide Vaccine (SurVaxM) in Patients with Recurrent Malignant Glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352.
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of Apoptosis and Mitotic Spindle Checkpoint by Survivin. Nature 1998, 396, 580–584.
- Ambrosini, G.; Adida, C.; Altieri, D.C. A Novel Anti-Apoptosis Gene, Survivin, Expressed in Cancer and Lymphoma. Nat. Med. 1997, 3, 917–921.
- Kajiwara, Y.; Yamasaki, F.; Hama, S.; Yahara, K.; Yoshioka, H.; Sugiyama, K.; Arita, K.; Kurisu, K. Expression of Survivin in Astrocytic Tumors: Correlation with Malignant Grade and Prognosis. Cancer 2003, 97, 1077–1083.
- Satoh, K.; Kaneko, K.; Hirota, M.; Masamune, A.; Satoh, A.; Shimosegawa, T. Expression of Survivin Is Correlated with Cancer Cell Apoptosis and Is Involved in the Development of Human Pancreatic Duct Cell Tumors. Cancer 2001, 92, 271–278.
- Galbo, P.M.; Ciesielski, M.J.; Figel, S.; Maguire, O.; Qiu, J.; Wiltsie, L.; Minderman, H.; Fenstermaker, R.A. Circulating CD9+/GFAP+/Survivin+ Exosomes in Malignant Glioma Patients Following Survivin Vaccination. Oncotarget 2017, 8, 114722–114735.
- MimiVax, LLC. Prospective Randomized Placebo-Controlled Trial of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma (SURVIVE). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05163080 (accessed on 28 February 2022).
- Chiocca, E.A.; Nassiri, F.; Wang, J.; Peruzzi, P.; Zadeh, G. Viral and Other Therapies for Recurrent Glioblastoma: Is a 24-Month Durable Response Unusual? Neuro-Oncology 2019, 21, 14–25.
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866.
- Rainov, N.G. A Phase III Clinical Evaluation of Herpes Simplex Virus Type 1 Thymidine Kinase and Ganciclovir Gene Therapy as an Adjuvant to Surgical Resection and Radiation in Adults with Previously Untreated Glioblastoma Multiforme. Hum. Gene Ther. 2000, 11, 2389–2401.
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated Multi-Mutated Herpes Simplex Virus-1 for the Treatment of Malignant Gliomas. Nat. Med. 1995, 1, 938–943.
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055.
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874.
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622.
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of RQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther.-Methods Clin. Dev. 2020, 17, 871–893.
- Chiocca, E.A. A Phase I Study of the Treatment of Recurrent Malignant Glioma with RQNestin34.5v.2, a Genetically Engineered HSV-1 Virus, and Immunomodulation with Cyclophosphamide. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03152318 (accessed on 28 February 2022).
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB Study of Gene-Mediated Cytotoxic Immunotherapy Adjuvant to up-Front Surgery and Intensive Timing Radiation for Malignant Glioma. J. Clin. Oncol. 2011, 29, 3611–3619.
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II Multicenter Study of Gene-Mediated Cytotoxic Immunotherapy as Adjuvant to Surgical Resection for Newly Diagnosed Malignant Glioma. Neuro-Oncology 2016, 18, 1137–1145.
- Westphal, M.; Ylä-Herttuala, S.; Martin, J.; Warnke, P.; Menei, P.; Eckland, D.; Kinley, J.; Kay, R.; Ram, Z.; ASPECT Study Group. Adenovirus-Mediated Gene Therapy with Sitimagene Ceradenovec Followed by Intravenous Ganciclovir for Patients with Operable High-Grade Glioma (ASPECT): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2013, 14, 823–833.
- Chiocca, E.A.; Smith, K.M.; McKinney, B.; Palmer, C.A.; Rosenfeld, S.; Lillehei, K.; Hamilton, A.; DeMasters, B.K.; Judy, K.; Kirn, D. A Phase I Trial of Ad.HIFN-β Gene Therapy for Glioma. Mol. Ther. 2008, 16, 618–626.
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161.
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal Ribosomal Entry Site Substitution Eliminates Neurovirulence in Intergeneric Poliovirus Recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375.
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR Plays a Key Role in Cell Motility during Tumor Cell Invasion and Migration. BMC Cancer 2004, 4, 73.
- Lupo, K.B.; Matosevic, S. CD155 Immunoregulation as a Target for Natural Killer Cell Immunotherapy in Glioblastoma. J. Hematol. Oncol. 2020, 13, 76.
- Brown, M.C.; Gromeier, M. Cytotoxic and Immunogenic Mechanisms of Recombinant Oncolytic Poliovirus. Curr. Opin. Virol. 2015, 13, 81–85.
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.-G.; Kiessling, R.; Malmberg, K.-J. Primary Human Tumor Cells Expressing CD155 Impair Tumor Targeting by Down-Regulating DNAM-1 on NK Cells. J. Immunol. 2009, 183, 4921–4930.
- Strauss, M.; Filman, D.J.; Belnap, D.M.; Cheng, N.; Noel, R.T.; Hogle, J.M. Nectin-like Interactions between Poliovirus and Its Receptor Trigger Conformational Changes Associated with Cell Entry. J. Virol. 2015, 89, 4143–4157.
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable Complete Responses in Some Recurrent High-Grade Glioma Patients Treated with Toca 511 + Toca FC. Neuro-Oncology 2018, 20, 1383–1392.
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination with Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients with Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946.
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106.
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging-Guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646.
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The Blood–Brain Barrier and Blood–Tumour Barrier in Brain Tumours and Metastases. Nat. Rev. Cancer 2020, 20, 26–41.
- Poon, C.; McMahon, D.; Hynynen, K. Noninvasive and Targeted Delivery of Therapeutics to the Brain Using Focused Ultrasound. Neuropharmacology 2017, 120, 20–37.
- Aryal, M.; Vykhodtseva, N.; Zhang, Y.-Z.; Park, J.; McDannold, N. Multiple Treatments with Liposomal Doxorubicin and Ultrasound-Induced Disruption of Blood-Tumor and Blood-Brain Barriers Improve Outcomes in a Rat Glioma Model. J. Control. Release 2013, 169, 103–111.
- McDannold, N.; Arvanitis, C.D.; Vykhodtseva, N.; Livingstone, M.S. Temporary Disruption of the Blood-Brain Barrier by Use of Ultrasound and Microbubbles: Safety and Efficacy Evaluation in Rhesus Macaques. Cancer Res. 2012, 72, 3652–3663.
- Lynch, M.; Heinen, S.; Markham-Coultes, K.; O’Reilly, M.; Van Slyke, P.; Dumont, D.J.; Hynynen, K.; Aubert, I. Vasculotide Restores the Blood-Brain Barrier after Focused Ultrasound-Induced Permeability in a Mouse Model of Alzheimer’s Disease. Int. J. Med. Sci. 2021, 18, 482–493.
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the Blood-Brain Barrier by Focused Ultrasound Induces Sterile Inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84.
- Shi, L.; Palacio-Mancheno, P.; Badami, J.; Shin, D.W.; Zeng, M.; Cardoso, L.; Tu, R.; Fu, B.M. Quantification of Transient Increase of the Blood-Brain Barrier Permeability to Macromolecules by Optimized Focused Ultrasound Combined with Microbubbles. Int. J. Nanomed. 2014, 9, 4437–4448.
- Marquet, F.; Tung, Y.-S.; Teichert, T.; Ferrera, V.P.; Konofagou, E.E. Noninvasive, Transient and Selective Blood-Brain Barrier Opening in Non-Human Primates In Vivo. PLoS ONE 2011, 6, e22598.
- McDannold, N.; Zhang, Y.; Supko, J.G.; Power, C.; Sun, T.; Vykhodtseva, N.; Golby, A.J.; Reardon, D.A. Blood-Brain Barrier Disruption and Delivery of Irinotecan in a Rat Model Using a Clinical Transcranial MRI-Guided Focused Ultrasound System. Sci. Rep. 2020, 10, 8766.
- Todd, N.; Angolano, C.; Ferran, C.; Devor, A.; Borsook, D.; McDannold, N. Secondary Effects on Brain Physiology Caused by Focused Ultrasound-Mediated Disruption of the Blood-Brain Barrier. J. Control. Release 2020, 324, 450–459.
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-Invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321.
- Abrahao, A.; Meng, Y.; Llinas, M.; Huang, Y.; Hamani, C.; Mainprize, T.; Aubert, I.; Heyn, C.; Black, S.E.; Hynynen, K.; et al. First-in-Human Trial of Blood–Brain Barrier Opening in Amyotrophic Lateral Sclerosis Using MR-Guided Focused Ultrasound. Nat. Commun. 2019, 10, 4373.
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical Trial of Blood-Brain Barrier Disruption by Pulsed Ultrasound. Sci. Transl. Med. 2016, 8, 343re2.
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood-Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801.
- Meng, Y.; MacIntosh, B.J.; Shirzadi, Z.; Kiss, A.; Bethune, A.; Heyn, C.; Mithani, K.; Hamani, C.; Black, S.E.; Hynynen, K.; et al. Resting State Functional Connectivity Changes after MR-Guided Focused Ultrasound Mediated Blood-Brain Barrier Opening in Patients with Alzheimer’s Disease. Neuroimage 2019, 200, 275–280.
- Rezai, A.R.; Ranjan, M.; D’Haese, P.-F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive Hippocampal Blood−brain Barrier Opening in Alzheimer’s Disease with Focused Ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182.
- Deng, Z.; Sheng, Z.; Yan, F. Ultrasound-Induced Blood-Brain-Barrier Opening Enhances Anticancer Efficacy in the Treatment of Glioblastoma: Current Status and Future Prospects. J. Oncol. 2019, 2019, e2345203.
- Alli, S.; Figueiredo, C.A.; Golbourn, B.; Sabha, N.; Wu, M.Y.; Bondoc, A.; Luck, A.; Coluccia, D.; Maslink, C.; Smith, C.; et al. Brainstem Blood Brain Barrier Disruption Using Focused Ultrasound: A Demonstration of Feasibility and Enhanced Doxorubicin Delivery. J. Control. Release 2018, 281, 29–41.
- Gasca-Salas, C.; Fernández-Rodríguez, B.; Pineda-Pardo, J.A.; Rodríguez-Rojas, R.; Obeso, I.; Hernández-Fernández, F.; del Álamo, M.; Mata, D.; Guida, P.; Ordás-Bandera, C.; et al. Blood-Brain Barrier Opening with Focused Ultrasound in Parkinson’s Disease Dementia. Nat. Commun. 2021, 12, 779.
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–Brain Barrier Opening in Alzheimer’s Disease Using MR-Guided Focused Ultrasound. Nat. Commun. 2018, 9, 2336.
- Pouliopoulos, A.N.; Kwon, N.; Jensen, G.; Meaney, A.; Niimi, Y.; Burgess, M.T.; Ji, R.; McLuckie, A.J.; Munoz, F.A.; Kamimura, H.A.S.; et al. Safety Evaluation of a Clinical Focused Ultrasound System for Neuronavigation Guided Blood-Brain Barrier Opening in Non-Human Primates. Sci. Rep. 2021, 11, 15043.
- Chen, K.-T.; Chai, W.-Y.; Lin, Y.-J.; Lin, C.-J.; Chen, P.-Y.; Tsai, H.-C.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-Guided Focused Ultrasound for Transcranial Blood-Brain Barrier Opening and Immunostimulation in Brain Tumors. Sci. Adv. 2021, 7, eabd0772.
- Conti, A.; Mériaux, S.; Larrat, B. About the Marty Model of Blood-Brain Barrier Closure after Its Disruption Using Focused Ultrasound. Phys. Med. Biol. 2019, 64, 14NT02.
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Effects of Acoustic Parameters and Ultrasound Contrast Agent Dose on Focused-Ultrasound Induced Blood-Brain Barrier Disruption. Ultrasound Med. Biol. 2008, 34, 930–937.
- Marty, B.; Larrat, B.; Van Landeghem, M.; Robic, C.; Robert, P.; Port, M.; Le Bihan, D.; Pernot, M.; Tanter, M.; Lethimonnier, F.; et al. Dynamic Study of Blood-Brain Barrier Closure after Its Disruption Using Ultrasound: A Quantitative Analysis. J. Cereb. Blood Flow Metab. 2012, 32, 1948–1958.
- Park, J.; Zhang, Y.; Vykhodtseva, N.; Jolesz, F.A.; McDannold, N.J. The Kinetics of Blood Brain Barrier Permeability and Targeted Doxorubicin Delivery into Brain Induced by Focused Ultrasound. J. Control. Release 2012, 162, 134–142.
- O’Reilly, M.A.; Waspe, A.C.; Ganguly, M.; Hynynen, K. Focused-Ultrasound Disruption of the Blood-Brain Barrier Using Closely-Timed Short Pulses: Influence of Sonication Parameters and Injection Rate. Ultrasound Med. Biol. 2011, 37, 587–594.
- Samiotaki, G.; Vlachos, F.; Tung, Y.-S.; Konofagou, E.E. A Quantitative Pressure and Microbubble-Size Dependence Study of Focused Ultrasound-Induced Blood-Brain Barrier Opening Reversibility in Vivo Using MRI. Magn. Reson. Med. 2012, 67, 769–777.
- Chen, K.-T.; Wei, K.-C.; Liu, H.-L. Theranostic Strategy of Focused Ultrasound Induced Blood-Brain Barrier Opening for CNS Disease Treatment. Front. Pharmacol. 2019, 10, 86.
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-Mediated Blood-Brain Barrier Disruption for Targeted Drug Delivery in the Central Nervous System. Adv. Drug Deliv. Rev. 2014, 72, 94–109.
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Use of Ultrasound Pulses Combined with Definity for Targeted Blood-Brain Barrier Disruption: A Feasibility Study. Ultrasound Med. Biol. 2007, 33, 584–590.
- Chu, P.-C.; Liu, H.-L.; Lai, H.-Y.; Lin, C.-Y.; Tsai, H.-C.; Pei, Y.-C. Neuromodulation Accompanying Focused Ultrasound-Induced Blood-Brain Barrier Opening. Sci. Rep. 2015, 5, 15477.
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Targeted Disruption of the Blood-Brain Barrier with Focused Ultrasound: Association with Cavitation Activity. Phys. Med. Biol. 2006, 51, 793–807.
- O’Reilly, M.A.; Hynynen, K. Blood-Brain Barrier: Real-Time Feedback-Controlled Focused Ultrasound Disruption by Using an Acoustic Emissions-Based Controller. Radiology 2012, 263, 96–106.
- Sun, T.; Samiotaki, G.; Wang, S.; Acosta, C.; Chen, C.C.; Konofagou, E.E. Acoustic Cavitation-Based Monitoring of the Reversibility and Permeability of Ultrasound-Induced Blood-Brain Barrier Opening. Phys. Med. Biol. 2015, 60, 9079–9094.
- Sun, T.; Zhang, Y.; Power, C.; Alexander, P.M.; Sutton, J.T.; Aryal, M.; Vykhodtseva, N.; Miller, E.L.; McDannold, N.J. Closed-Loop Control of Targeted Ultrasound Drug Delivery across the Blood–Brain/Tumor Barriers in a Rat Glioma Model. Proc. Natl. Acad. Sci. USA 2017, 114, E10281–E10290.
- Patel, A.; Schoen, S.J.; Arvanitis, C.D. Closed-Loop Spatial and Temporal Control of Cavitation Activity with Passive Acoustic Mapping. IEEE Trans. Biomed. Eng. 2019, 66, 2022–2031.
- Collis, J.; Manasseh, R.; Liovic, P.; Tho, P.; Ooi, A.; Petkovic-Duran, K.; Zhu, Y. Cavitation Microstreaming and Stress Fields Created by Microbubbles. Ultrasonics 2010, 50, 273–279.
- Sutton, J.T.; Haworth, K.J.; Pyne-Geithman, G.; Holland, C.K. Ultrasound-Mediated Drug Delivery for Cardiovascular Disease. Expert Opin. Drug Deliv. 2013, 10, 573–592.
- Guo, Y.; Lee, H.; Fang, Z.; Velalopoulou, A.; Kim, J.; Ben Thomas, M.; Liu, J.; Abramowitz, R.G.; Kim, Y.; Coskun, A.F.; et al. Single-Cell Analysis Reveals Effective SiRNA Delivery in Brain Tumors with Microbubble-Enhanced Ultrasound and Cationic Nanoparticles. Sci. Adv. 2021, 7, eabf7390.
- Anastasiadis, P.; Gandhi, D.; Guo, Y.; Ahmed, A.-K.; Bentzen, S.M.; Arvanitis, C.; Woodworth, G.F. Localized Blood–Brain Barrier Opening in Infiltrating Gliomas with MRI-Guided Acoustic Emissions–Controlled Focused Ultrasound. Proc. Natl. Acad. Sci. USA 2021, 118, e2103280118.
- Liu, H.-L.; Hsu, P.-H.; Lin, C.-Y.; Huang, C.-W.; Chai, W.-Y.; Chu, P.-C.; Huang, C.-Y.; Chen, P.-Y.; Yang, L.-Y.; Kuo, J.S.; et al. Focused Ultrasound Enhances Central Nervous System Delivery of Bevacizumab for Malignant Glioma Treatment. Radiology 2016, 281, 99–108.
- Coluccia, D.; Figueiredo, C.A.; Wu, M.Y.; Riemenschneider, A.N.; Diaz, R.; Luck, A.; Smith, C.; Das, S.; Ackerley, C.; O’Reilly, M.; et al. Enhancing Glioblastoma Treatment Using Cisplatin-Gold-Nanoparticle Conjugates and Targeted Delivery with Magnetic Resonance-Guided Focused Ultrasound. Nanomedicine 2018, 14, 1137–1148.
- Thévenot, E.; Jordão, J.F.; O’Reilly, M.A.; Markham, K.; Weng, Y.-Q.; Foust, K.D.; Kaspar, B.K.; Hynynen, K.; Aubert, I. Targeted Delivery of Self-Complementary Adeno-Associated Virus Serotype 9 to the Brain, Using Magnetic Resonance Imaging-Guided Focused Ultrasound. Hum. Gene Ther. 2012, 23, 1144–1155.
- Noroozian, Z.; Xhima, K.; Huang, Y.; Kaspar, B.K.; Kügler, S.; Hynynen, K.; Aubert, I. MRI-Guided Focused Ultrasound for Targeted Delivery of RAAV to the Brain. Methods Mol. Biol. 2019, 1950, 177–197.
- Jordão, J.F.; Ayala-Grosso, C.A.; Markham, K.; Huang, Y.; Chopra, R.; McLaurin, J.; Hynynen, K.; Aubert, I. Antibodies Targeted to the Brain with Image-Guided Focused Ultrasound Reduces Amyloid-Beta Plaque Load in the TgCRND8 Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e10549.
- Kobus, T.; Zervantonakis, I.K.; Zhang, Y.; McDannold, N.J. Growth Inhibition in a Brain Metastasis Model by Antibody Delivery Using Focused Ultrasound-Mediated Blood-Brain Barrier Disruption. J. Control. Release 2016, 238, 281–288.
- Morse, S.V.; Mishra, A.; Chan, T.G.; de Rosales, R.M.; Choi, J.J.; Choi, J.J. Liposome Delivery to the Brain with Rapid Short-Pulses of Focused Ultrasound and Microbubbles. J. Control. Release 2022, 341, 605–615.
- Chan, T.G.; Morse, S.V.; Copping, M.J.; Choi, J.J.; Vilar, R. Targeted Delivery of DNA-Au Nanoparticles across the Blood-Brain Barrier Using Focused Ultrasound. ChemMedChem 2018, 13, 1311–1314.
- Zhao, G.; Huang, Q.; Wang, F.; Zhang, X.; Hu, J.; Tan, Y.; Huang, N.; Wang, Z.; Wang, Z.; Cheng, Y. Targeted ShRNA-Loaded Liposome Complex Combined with Focused Ultrasound for Blood Brain Barrier Disruption and Suppressing Glioma Growth. Cancer Lett. 2018, 418, 147–158.
- Burgess, A.; Ayala-Grosso, C.A.; Ganguly, M.; Jordão, J.F.; Aubert, I.; Hynynen, K. Targeted Delivery of Neural Stem Cells to the Brain Using MRI-Guided Focused Ultrasound to Disrupt the Blood-Brain Barrier. PLoS ONE 2011, 6, e27877.
- Alkins, R.; Burgess, A.; Kerbel, R.; Wels, W.S.; Hynynen, K. Early Treatment of HER2-Amplified Brain Tumors with Targeted NK-92 Cells and Focused Ultrasound Improves Survival. Neuro-Oncology 2016, 18, 974–981.
- McDannold, N.; Zhang, Y.; Supko, J.G.; Power, C.; Sun, T.; Peng, C.; Vykhodtseva, N.; Golby, A.J.; Reardon, D.A. Acoustic Feedback Enables Safe and Reliable Carboplatin Delivery across the Blood-Brain Barrier with a Clinical Focused Ultrasound System and Improves Survival in a Rat Glioma Model. Theranostics 2019, 9, 6284–6299.
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic Targeting of Tumor-Associated Myeloid Cells Synergizes with Radiation Therapy for Glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723.
- Taiarol, L.; Formicola, B.; Magro, R.D.; Sesana, S.; Re, F. An Update of Nanoparticle-Based Approaches for Glioblastoma Multiforme Immunotherapy. Nanomedicine 2020, 15, 1861–1871.
- Mathew, E.N.; Berry, B.C.; Yang, H.W.; Carroll, R.S.; Johnson, M.D. Delivering Therapeutics to Glioblastoma: Overcoming Biological Constraints. Int. J. Mol. Sci. 2022, 23, 1711.
- Zhang, F.; Stephan, S.B.; Ene, C.I.; Smith, T.T.; Holland, E.C.; Stephan, M.T. Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-Cell Therapy in Solid Malignancies. Cancer Res. 2018, 78, 3718–3730.
- Beccaria, K.; Sabbagh, A.; de Groot, J.; Canney, M.; Carpentier, A.; Heimberger, A.B. Blood-Brain Barrier Opening with Low Intensity Pulsed Ultrasound for Immune Modulation and Immune Therapeutic Delivery to CNS Tumors. J. Neuro-Oncol. 2021, 151, 65–73.
- Chen, P.-Y.; Hsieh, H.-Y.; Huang, C.-Y.; Lin, C.-Y.; Wei, K.-C.; Liu, H.-L. Focused Ultrasound-Induced Blood–Brain Barrier Opening to Enhance Interleukin-12 Delivery for Brain Tumor Immunotherapy: A Preclinical Feasibility Study. J. Transl. Med. 2015, 13, 93.
- Curley, C.T.; Sheybani, N.D.; Bullock, T.N.; Price, R.J. Focused Ultrasound Immunotherapy for Central Nervous System Pathologies: Challenges and Opportunities. Theranostics 2017, 7, 3608–3623.
- Sevenich, L. Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front. Oncol. 2019, 9, 163.
- Steeg, P.S. The Blood-Tumour Barrier in Cancer Biology and Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 696–714.
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, K.; et al. Opening of the Blood-Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337.
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused Ultrasound Delivers Targeted Immune Cells to Metastatic Brain Tumors. Cancer Res. 2013, 73, 1892–1899.
- Pacia, C.P.; Yuan, J.; Yue, Y.; Xu, L.; Nazeri, A.; Desai, R.; Gach, H.M.; Wang, X.; Talcott, M.R.; Chaudhuri, A.A.; et al. Sonobiopsy for Minimally Invasive, Spatiotemporally-Controlled, and Sensitive Detection of Glioblastoma-Derived Circulating Tumor DNA. Theranostics 2022, 12, 362–378.
- Yuan, J.; Ye, D.; Chen, S.; Chen, H. Therapeutic Ultrasound-Enhanced Immune Checkpoint Inhibitor Therapy. Front. Phys. 2021, 9, 102.
- Elias, W.J.; Huss, D.; Voss, T.; Loomba, J.; Khaled, M.; Zadicario, E.; Frysinger, R.C.; Sperling, S.A.; Wylie, S.; Monteith, S.J.; et al. A Pilot Study of Focused Ultrasound Thalamotomy for Essential Tremor. N. Engl. J. Med. 2013, 369, 640–648.
- Elias, W.J.; Lipsman, N.; Ondo, W.G.; Ghanouni, P.; Kim, Y.G.; Lee, W.; Schwartz, M.; Hynynen, K.; Lozano, A.M.; Shah, B.B.; et al. A Randomized Trial of Focused Ultrasound Thalamotomy for Essential Tremor. N. Engl. J. Med. 2016, 375, 730–739.
- Iorio-Morin, C.; Yamamoto, K.; Sarica, C.; Zemmar, A.; Levesque, M.; Brisebois, S.; Germann, J.; Loh, A.; Boutet, A.; Elias, G.J.B.; et al. Bilateral Focused Ultrasound Thalamotomy for Essential Tremor (BEST-FUS Phase 2 Trial). Mov. Disord. 2021, 36, 2653–2662.
- Martin, E.; Jeanmonod, D.; Morel, A.; Zadicario, E.; Werner, B. High-Intensity Focused Ultrasound for Noninvasive Functional Neurosurgery. Ann. Neurol. 2009, 66, 858–861.
- Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Kheirolomoom, A.; Liu, Y.; Fite, B.Z.; Tam, S.M.; Tucci, S.T.; Watson, K.D.; Wong, A.W.; et al. Priming Is Key to Effective Incorporation of Image-Guided Thermal Ablation into Immunotherapy Protocols. JCI Insight 2017, 2, e90521.
- Chavez, M.; Silvestrini, M.T.; Ingham, E.S.; Fite, B.Z.; Mahakian, L.M.; Tam, S.M.; Ilovitsh, A.; Monjazeb, A.M.; Murphy, W.J.; Hubbard, N.E.; et al. Distinct Immune Signatures in Directly Treated and Distant Tumors Result from TLR Adjuvants and Focal Ablation. Theranostics 2018, 8, 3611–3628.
- Eranki, A.; Srinivasan, P.; Ries, M.; Kim, A.; Lazarski, C.A.; Rossi, C.T.; Khokhlova, T.D.; Wilson, E.; Knoblach, S.M.; Sharma, K.V.; et al. High-Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2020, 26, 1152–1161.
- Qu, S.; Worlikar, T.; Felsted, A.E.; Ganguly, A.; Beems, M.V.; Hubbard, R.; Pepple, A.L.; Kevelin, A.A.; Garavaglia, H.; Dib, J.; et al. Non-Thermal Histotripsy Tumor Ablation Promotes Abscopal Immune Responses That Enhance Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e000200.
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High Intensity Focused Ultrasound: The Fundamentals, Clinical Applications and Research Trends. Diagn. Interv. Imaging 2018, 99, 349–359.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
772
Revisions:
2 times
(View History)
Update Date:
18 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No