Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christophe Ravaud | -- | 5192 | 2022-04-12 17:21:19 | | | |
| 2 | Vivi Li | -43 word(s) | 5149 | 2022-04-13 04:36:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ravaud, C.; Ved, N.; Jackson, D.G.; Vieira, J.M.; Riley, P.R. Lymphatic Clearance of Immune Cells in Cardiovascular Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/21665 (accessed on 24 July 2026).
Ravaud C, Ved N, Jackson DG, Vieira JM, Riley PR. Lymphatic Clearance of Immune Cells in Cardiovascular Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/21665. Accessed July 24, 2026.
Ravaud, Christophe, Nikita Ved, David G. Jackson, Joaquim Miguel Vieira, Paul R. Riley. "Lymphatic Clearance of Immune Cells in Cardiovascular Disease" Encyclopedia, https://encyclopedia.pub/entry/21665 (accessed July 24, 2026).
Ravaud, C., Ved, N., Jackson, D.G., Vieira, J.M., & Riley, P.R. (2022, April 12). Lymphatic Clearance of Immune Cells in Cardiovascular Disease. In Encyclopedia. https://encyclopedia.pub/entry/21665
Ravaud, Christophe, et al. "Lymphatic Clearance of Immune Cells in Cardiovascular Disease." Encyclopedia. Web. 12 April, 2022.
Copy Citation
The lymphatic vasculature is a vital component of the cardiovascular system, consisting of a blind-ended, highly permeable vascular network, integral in maintaining tissue homeostasis, regulation of interstitial fluid, lipid absorption, fluid drainage, and immune cell trafficking. Its role in immune cell transport is critical in the initiation of the immune response, especially following injury. This is of particular importance in the heart, where the lymphatic vasculature plays a vital role in myocardial healing following cardiac injury. By promoting cell egress or exit from the heart, the lymphatic systems favour cell clearance by way of reduction of the immune cell load in damaged tissue.
lymphangiogenesis
myocardial infarction
immune cells
lymphatic
cell clearance
VEGF-C
LYVE1
1. Immune Cells in the Heart
1.1. Neutrophils: The First Cells to Arrive at the Site of Infarction
Within a few hours post-MI, the necrosis of cardiomyocytes and the consequent release of cell debris generates endogenous agents known as damage-associated molecular patterns (DAMPs) [1][2][3] which activate and recruit neutrophils to the site of infarction via their Toll-like receptors [4][5]. Neutrophils are defined as CD45+CD11b+Ly6C+ cells, but different subsets exist. As with other immune cells such as macrophages, neutrophils can be subcategorised into N1 and N2 subpopulations depending on their pro-inflammatory or anti-inflammatory profiles respectively [4]. At an early-stage post-MI (Day 1), N1 neutrophils (Ly6G+CD206−) exhibit high expression of pro-inflammatory markers, generate high levels of reactive oxygen species and initiate local inflammation as well as tissue destruction. By phagocytosis, they clear debris and dead cells in the infarcted myocardium [6]. From Day 5 post-MI, an N2 neutrophil population (Ly6G+CD206+), with an anti-inflammatory phenotype, begins to increase and plays an important role in the resolution of the infarct wound [4]. These complementary but opposing roles for neutrophils post-MI are necessary for heart repair and must be tightly regulated. Neutrophil depletion in a mouse model of MI worsens cardiac function and increases fibrosis. Interestingly, in the absence of neutrophils, the number of monocytes recruited to the infarct zone is reduced, whereas the number of macrophages is increased [7]. Moreover, the neutrophil gelatinase-associated protein lipocalin, secreted by neutrophils, polarises macrophages towards a reparative phenotype [7]. Paradoxically, the absence of neutrophils is deleterious; however, elevated neutrophil counts in ST-segment elevation myocardial infarction (STEMI) show a positive correlation with myocardial infarct size [8]. Thus, timely clearance of neutrophils from the infarct zone, may present a tractable therapeutic approach to improve heart recovery post-MI.
1.2. Monocyte/Macrophages: Cells Involved in Both Inflammation and Its Resolution
Monocytes and macrophages dominate the innate immune response post-MI in terms of sheer numbers and are the best studied leucocyte population in this context to date. Macrophages can be broadly categorised as either tissue-resident cells that emerge during embryonic development or monocyte-derived cells that derive from bone marrow and splenic reservoirs [9]. Tissue-resident macrophages originate from the yolk sac or foetal liver progenitors [10][11] and represent up to 8% of the total non-cardiomyocyte population in the healthy adult mouse heart [12]. In the embryonic mouse heart, they mediate remodelling of the coronary plexus and lymphatic endothelium, and thus appear as indispensable for development of the mammalian cardiac vasculature [13][14]. In adults, their primary role is to maintain homeostasis of the myocardium in the steady-state heart by removing senescent and dying cells and facilitating electrical conduction [15]. In addition, they are involved in the first steps of the inflammatory response following MI. Through a myeloid differentiation primary response-88 (MYD88)-dependent pathway, tissue-resident C-C motif chemokine receptor 2-positive (CCR2+) macrophages are responsible for the initial recruitment of monocytes to the infarct zone and the promotion of neutrophil extravasation [16][17]. Even if the resident macrophages are rapidly replaced by circulating monocytes, their genetic deletion decreases cardiac output and contributes to adverse remodelling, underlying their crucial cardioprotective role [18]. During the first phase of inflammation, high levels of monocyte chemoattractant protein-1(MCP-1, a CCR2 ligand) in the myocardium triggers the infiltration and accumulation of Ly-6Chigh monocytes into the damaged tissue from the first day post-injury [19]. Ly-6Chigh monocytes peak between Day 3 and Day 5 post-MI, and display high phagocytic and proteolytic activities, and express proinflammatory cytokines including tumour necrosis factor-α (TNFα) [20]. Genetic deletion of Ly-6Chigh monocytes in Ccr2−/− mice impairs cardiac function and leads to higher elastin levels in the fibrotic scar tissue [21]. From Day 5 onwards, Ly-6Clow monocytes accumulate and trigger reparative processes including angiogenesis, by expressing high levels of vascular endothelial growth factor (VEGF), and inducing deposition of extracellular matrix (ECM) [20]. Up-regulation of macrophage-colony stimulating factor (MCSF) in the pro-inflammatory environment of the myocardium induces monocyte differentiation into macrophages [22].
In the mouse, macrophages differ from monocytes by virtue of increased F4/80 expression, while in the human they display increased expression of CD68 and the Major Histocompatibility Complex II (MHC II) and decreased expression of CD14 [23]. As for monocytes, two main subsets of macrophages occur in both species post-MI. Historically, macrophages have been divided into pro-inflammatory (M1) and anti-inflammatory or pro-resolving (M2); however, the reality is more complex as there is great heterogeneity in macrophage populations post-MI [24]. The early stage of MI resolution is characterised by the presence of pro-inflammatory M1 macrophages to promote clearance of matrix and debris through phagocytosis. Those pro-inflammatory macrophages produce various cytokines including interleukin (IL) -1β (IL-1β), IL6, and TNFα to sustain the inflammatory environment and to activate resident fibroblasts. As a result, several matrix metalloproteinases (MMPs) are released into the micro-environment and induce further ECM degradation [25]. This process is essential and must be tightly regulated to ensure an optimum environment before the resolution phase. The absence of this step or a prolonged inflammatory response leads to extensive damage and poor healing [26][27]. As M2 macrophages are reparative, they express anti-inflammatory cytokines (e.g., IL10). By secreting important growth factors, such as VEGF and transforming growth factor -β (TGF-β), they promote cell proliferation, angiogenesis, and ECM production respectively. TGF-β controls the expression of alpha smooth muscle actin (α-SMA) in the resident fibroblasts which drives their differentiation into myofibroblasts [28]. Myofibroblasts are the primary source of ECM proteins to replace myocyte loss and form a reparative scar [29]. However, it has been shown recently that macrophages themselves can also contribute to collagen deposition, and thus also fibrosis, during heart repair [30]. Deficiency of the pseudokinase Tribbles homolog 1 (Trib1) impairs the ability to form M2 macrophages without affecting the other immune cells [31]. Trib1−/− mice, which exhibit a selective depletion of M2-like macrophages, present a disastrous prognosis following MI. These mice display regular cardiac rupture due to reduced collagen fibril formation and by extension, poor infarct repair [32]. In contrast, depleting cardiac inflammatory monocytes and shifting macrophages towards the anti-inflammatory phenotype in the heart post-MI enhances cardiac recovery and plays a cardioprotective role [33]. Similarly, inducing an M2 phenotype in the infarcted heart through administration of IL-4 enhances cardiac function in conjunction with diminished infarct size and intensified tissue repair. This presented as reinforced connective tissue structure formation, enhanced microvascular growth, and impaired cardiomyocyte hypertrophy [34]. Many other approaches, including transplantation with human umbilical cord blood mesenchymal stem cells [35] or injection of exosomes from adipose-derived mesenchymal stem cells [36], have been used to promote M2 macrophage polarisation post-MI and resulted in improved cardiac tissue repair in MI models (see review [37]).
1.3. Dendritic Cells: Regulators of Immune Tolerance during MI
Dendritic cells (DC) play an important role at the intersection of the innate and adaptive immune systems in injured tissues including the heart [38]. They are the most efficient antigen-presenting cells, and their main function is to activate naïve lymphocytes. In common with monocytes and macrophages, they express high levels of MHC II and CD11c. In mice, DCs infiltrate the heart from day 1 post-MI, peak at day 5 [1], and subsequently migrate to the mediastinal lymph nodes (MLNs) where they can activate T-lymphocytes [39]. A recent study also shows that a cross-priming DC population is present and activated in the heart following MI. This population activates cytotoxic CD8+ T cells and their deletion reduces myocardial immunopathology and functional decline [40]. DCs can be classified into two major subpopulations: conventional DC (cDC) and plasmacytoid DC (pDC). Interestingly, selective deletion of cDC, using the zinc finger and BTB domain containing 46 (Zbtb46) promoter [41] in mice post-MI, improves cardiac function and prevents adverse cardiac remodelling. This is accompanied by a decrease in fibrosis in the infarcted zone and an attenuation of the inflammatory state due to the reduction of the number of immune cells including macrophages, neutrophils, and T cells [42]. In contrast, selective deletion of pDC, using the specific promoter Bdca2 [43], does not affect heart function post-MI [42]. Total depletion of DCs promotes inflammation of the heart by increasing the infiltration of Ly-6Chigh monocytes and M1 macrophages and impairing the recruitment of Ly-6Clow monocytes and M2 macrophages to the infarcted myocardium. As a result, deterioration of left ventricular function and remodelling was observed [44]. In line with these observations, the presence of small numbers of DCs in infarcted myocardial tissue in humans is associated with increased macrophage infiltration, impaired reparative fibrosis, and an increased risk of cardiac rupture post-MI [45]. By contrast, injections of tolerogenic DCs (toDC) in mice 24 h and 7 days post-MI induces infarct-specific regulatory T cells (Treg) in the mediastinal lymph node and promotes an inflammatory-to-reparative macrophage shift in the myocardium [46]. Moreover, generation of toDCs with IL-37 and troponin 1 increases the number of Tregs in vitro and in the spleen in vivo. Their injection attenuates the infiltration of inflammatory cells in the infarcted hearts [47], decreases myocardial fibrosis, and improves cardiac function and survival post-MI [46][47].
Taken together, these studies underline the protective role of DCs in post-MI inflammation and as a result, the healing process. It is important to highlight that the beneficial role of DCs occurs through the recruitment and activation of other cells outside the injury zone (e.g., within the MLNs and spleen) and consequently, the lymphatic network may play a crucial role in this process.
2. Cardiac Lymphatic Network
2.1. Lymphatic Development in the Embryo
Under steady-state conditions the lymphatic system functions as normal. However, under pathological conditions, the lymphatic vasculature undergoes remodelling akin to what is seen during development. Therefore, understanding the development of the cardiac lymphatic system is essential in understanding its role in health and disease, and has previously been reviewed [48]. The lymphatic network arises both by budding from the blood vasculature and independently from lymphangioblasts during embryogenesis, in a process that is highly conserved in vertebrates. In the first instance, blood vessels are formed de novo through vasculogenesis from mesoderm- and somite-derived progenitors to form a primitive vascular network, which then matures through angiogenesis [49]. Subsequent to the formation of this vascular network, between embryonic days 9.5–10.5 (E9.5–E10.5) in mice, endothelial cells (ECs) in the cardinal vein (CV) then begin to express the lymphatic marker and master regulator of lymphatic endothelial cell specification Prospero-related homeobox domain 1 (PROX1) [50][51]. It is now clear that PROX1 is essential for the specification and ongoing development of the lymphatics, as Prox1 KO mice lack lymph sacs and lymphatic vessels [50] and show embryonic lethality. Furthermore, the endothelial-specific knockout of Prox1 results in lymphatic defects and postnatal lethality [52] and Prox1 overexpression is sufficient to direct ECs towards a lymphatic fate both in vitro [53] and in vivo [54], further emphasising its importance in lymphatic development. The PROX1+ve lymphatic endothelial cells (LECs) then bud from the cardinal vein and form primitive lymph sacs in response to the binding of VEGF-C [55] to its cognate, lymphatic endothelium-specific receptor, VEGF receptor 3 (VEGFR3). Vegf-c KO mice lack lymphatic vessels [55] and show embryonic lethality. Furthermore, Vegf-c+/− mice develop cutaneous lymphatic hypoplasia and lymphoedema, which can be rescued by VEGF-C treatment [55]. The nascent lymphatic network undergoes branching and network formation throughout later stages of development and postnatally, regulated by angiopoietin 2 (Ang2) and its receptor, TIE2. Ang2 KO mice show defects in lymphatic vascular remodelling [51], indicating that Ang2 may not be necessary for lymphangiogenesis de novo, but rather is critical for subsequent branching and pruning of the lymphatic vascular network.
In mice, a population of cardiac PROX1 and VEGFR3 expressing LECs originate from extra-cardiac tissue and the CV at E10.5 and migrate to the outflow tract and sinus venosus by E12.5, where they expand to form the cardiac lymphatics [56]. Much like the systemic lymphatics, the cardiac lymphatics follow the lead of the blood vascular system, and only appear following the formation of the sinus venosus, but before the onset of the coronary circulation [56]. Interestingly, the cardiac lymphatic vessels display significant heterogeneity, as lineage tracing identified that only 80% of cardiac LECs are venous-derived [56] with the remaining non-venous derived LECs, known as lymphangioblasts, originating from other sources including the haemogenic endothelium and are initially PROX1 deficient [57]. By E14.5, LECs mature and express lymphatic lineage markers including the LYmphatic Vessel Endothelial hyaluronan receptor 1 (LYVE-1) and the sialoglycoprotein podoplanin, by which point the cardiac lymphatics progressively extend from the base towards the apex of the heart, covering most of the developing organ [56][57]. Cardiac lymphangiogenesis is also dependent on VEGF-C and VEGF-D [55]. Transgenic mice expressing soluble VEGFR3 showed perturbed lymphatic development in the heart and other organs and also developed severe oedema and pericardial fluid accumulation [58]. Given that the epicardium and outflow tract is also a source of VEGF-C, they may be orchestrators of VEGF-C signalling and lymphatic sprouting [59]. Cardiac lymphatic vessel maturation and remodelling in mice continues postnatally for the following 2–3 weeks after birth [56][60]. Once fully matured, in humans, the lymphatic vasculature reaches all layers of the heart, including the atria, ventricles, and mitral valves [61]. In mice, the cardiac lymphatics extend predominantly around the branches of the coronary arteries and veins [62][63] and reside predominantly within the outer myocardium and compact wall of the chambers. Histological analysis at multiple developmental time points revealed that there are two pre-collector vessels. The left major pre-collector vessel runs along the left conal vein and under the left auricle towards the nearest lymph nodes. The second pre-collector runs parallel to the left cardiac vein toward the coronary sinus where extracardiac larger collector vessels empty into draining mediastinal lymph nodes which are found beneath the aortic arch and around the trachea [61][64]. Interestingly, mouse and human cardiac pre-collectors, unlike those of most other tissues contain very few smooth muscle cells [65][66]; thus, lymph propulsion from the heart appears to be dependent on extrinsic factors such as cardiac muscle contraction. Therefore, instances where cardiac contractility or heart rate are compromised, affect cardiac lymph flow accordingly [67][68].
2.2. Cardiac Lymphatic Remodelling Following Myocardial Infarction
Lymphatic remodelling is a process typically seen only during embryonic development or following injury or disease. In the heart, lymphatic remodelling can occur in both acute and chronic heart failure [58][69][70], following cardiac transplantation [71], and in atherosclerosis [72]. In recent years, research into enhancing lymphangiogenesis in the context of heart disease has become a key area of clinical interest. Cardiac lymphatic remodelling is often investigated in the context of MI in mice. One of the myriad consequences of MI and prolonged ischaemia is the death of endothelia, owing to increased vascular permeability and loss of lymphatic vessels, both of which result in poor fluid drainage and prolonged oedema [66][73]. In mammals, this loss of cardiovascular tissue results in heart remodelling, such that the dead myocardium is replaced with scar tissue. In an attempt to resolve the oedema, the cardiac lymphatics also undergo remodelling in the form of lymphangiogenesis in the infarct zone [20][66][74][75] (Figure 1). In parallel, following MI, there is an increase in local inflammatory cytokine and chemokine production, resulting in increased activation and recruitment of innate immune cells to the infarct area as discussed above [76]. Inadequate lymphangiogenesis results in increased interstitial fluid and osmotic pressures, which further amplify local immune responses through activation of osmoregulatory mechanisms in resident immune cells [77]. Moreover, inadequate lymphangiogenesis also results in the persistence of immune cells in the infarct region, as a result of their reduced clearance to draining cardiac lymph nodes. Ultimately, the events described result in fibrosis, impaired heart function, and eventually heart failure [67][73]. Furthermore, during the chronic phase following MI, the lymphatic remodelling of pre-collectors results in poor cardiac lymph transport leading to chronic myocardial oedema [26][56][66].
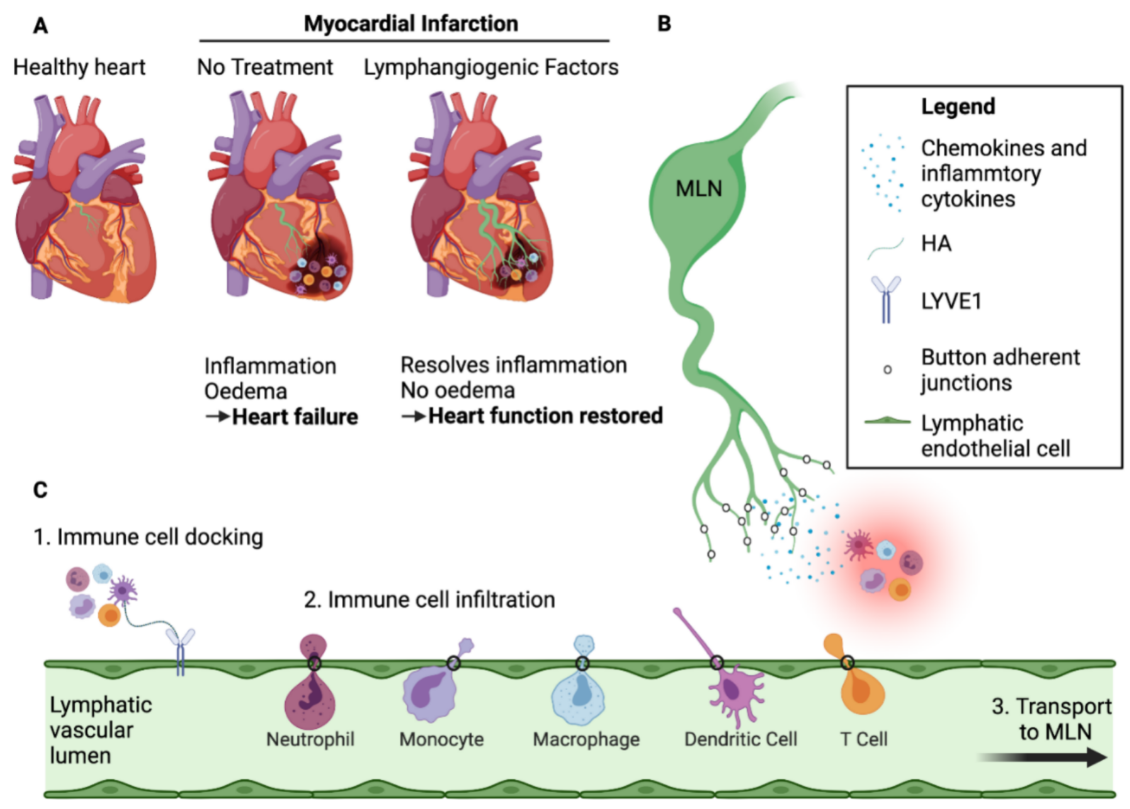
Figure 1. Lymphangiogenesis improves cardiac function post-MI. (A) Schematic comparison between a healthy heart and a heart subject to myocardial infarction (MI) with or without lymphangiogenic growth factor treatment. Following MI, immune cells infiltrate the infarct zone and create an inflammatory environment that impedes healing of the injured heart. Lymphangiogenesis occurs in non-treated hearts due to endogenous VEGF-C signaling, but is not sufficient to clear the immune cells and avoid myocardial oedema, resulting in scar formation and heart remodelling. Treatment with lymphangiogenic factors, such as VEGF-C(C156S) which binds exclusively to VEGFR3, greatly augments the lymphangiogenic response leading to efficient clearing of excess tissue fluid and pro-inflammatory cells, resulting in less severe heart remodelling and improved cardiac healing and function. (B) Cross-talk between lymphatic capillaries and immune cells in the infarcted area. Lymphatic endothelial cells secrete chemokines including CCL21, C-X3-C motif chemokine ligand 1(CX3CL1), and CXCL12, which attract specific populations of immune cells according to their receptor expression profile. These endothelial cells also express adhesion molecules, such as ICAM-1 and VCAM-1, which facilitate immune cell crawling. (C) Steps involved in immune cell clearance by the lymphatic vessels. Through their assembly of an endogenous HA surface glycocalyx, immune cells dock with LYVE-1 homodimers in the button-like endothelial junctions of initial afferent capillaries and enter the vessel lumen to migrate towards downstream MLNs. Created with BioRender.com.
2.3. Exit of Immune Cells from the Infarcted Heart—The Role of Cardiac Lymphatics
In addition to regulating interstitial fluid homeostasis, the lymphatics are an integral component of the immune system, facilitating the transport of immune cells, pathogens, and antigens [78] from the sites of injured and infected tissues to draining lymph nodes (dLNs) for generation of protective T and B cell responses [78][79]. Allied to this, they also provide a key route for exit of immune cells during the resolution of tissue inflammation, not only following myocardial infarction, but also lung injury and allograft rejection [74][80][81][82]. In normal resting tissue the numbers of immune cells migrating in afferent lymphatics are small, comprising mainly T-cells (approximately 90%) and immature antigen presenting dendritic cells (DCs) engaged in background immune surveillance. However, in injury and inflammation the numbers of immune cells in afferent lymph rise several-fold, due to an upsurge in lymph flow, an increase in lymph vessel permeability and the local release of pro-inflammatory cytokines [83]. The migrating populations include recirculating antigen-experienced memory T-cells (TRCM), immunoregulatory T-cells (Treg) and small numbers of B-cells that patrol the tissues for cognate antigen, mature DCs ferrying internalised antigens for immune priming, and macrophages and neutrophils involved in pathogen killing, clearance of tissue debris and tissue repair/remodelling [74][84][85][86][87]. The trafficking of each cell population is carefully choreographed, with antigen-charged DCs normally being the first to enter the lymphatics from the tissues. However, neutrophils are the most rapidly mobilised immune cells, and in some contexts (e.g., post-vaccination) the first to migrate to dLNs in inflammation, arriving some 72 h before either DCs or macrophages [88][89]. Of note, afferent lymph contains few if any naïve T or B cells, as these are absent from resting tissues. Instead, they enter LNs directly from the blood via high endothelial venules (HEVs) and recirculate via efferent lymph through the thoracic duct and subclavian veins [90].
The initial afferent vessels through which the above-mentioned immune cells enter the tissue lymphatics begin as blind-ended capillaries with a distinctive architecture composed of oakleaf-shaped LECs joined together by loose, discontinuous junctions that lack a substantial basement membrane [91]—characteristics well suited to a role in fluid drainage. Such junctions operate as primary valves that allow the one-way entry of fluid to the vessels while preventing its backflow to the interstitium [92][93]. Importantly, the interdigitating arrangement of oakleaf shaped endothelial cells creates a succession of overlapping flaps, and these are buttoned at their sides by adherens junction and tight junction proteins including VE-cadherin, JAMs, claudins and ESAM, while more loosely attached at their tips by the homotypic Platelet Endothelial Cell Adhesion Molecule, PECAM-1 (a.k.a. CD31), and the HA receptor LYVE-1 [94]. Notably, as revealed by electron microscopy and high-resolution confocal imaging, the alternating flaps guard openings of ~0.5–1µm in size [95] that act as portals for migrating DCs and macrophages which enter the afferent lymphatics by a process of pushing and squeezing [95][96][97]. Moreover, as discussed below, the discrete location of LYVE-1 at these portals is fully consistent with the key function of the receptor in mediating such immune cell entry.
Downstream of initial lymphatics, the larger pre-collector and collector vessels are more tightly sealed by conventional tight “zipper”-like junctions, akin to those of blood vessels. In keeping, their constituent LECs have a more regular rather than oakleaf shape and express far lower levels of LYVE-1 [94][95]. In addition, the pre-collector/collectors are invested by smooth muscle cells, whose contraction enables the conveyance of leukocytes to dLNs via lymph flow [98]. Notably, lymphatic vessels present in embryonic tissue have exclusively zippered junctions, and only transition to the button-like junctions of initial capillaries late in development and during the early neonatal period. Moreover, in chronic inflammation and tissue injury, the lymphatics also display significant junctional plasticity, such that new vessels generated during lymphangiogenesis as well as surrounding pre-existing vessels have zipper-like junctions similar to those of early embryos [94].
3. Lymphangiogenic Therapy Post-MI
It is well known from studies in both humans and rodents that MI induces pathological remodelling of the cardiac lymphatics [56][66][69][99][100]. Indeed, the characteristic oedema in the myocardial interstitium that results from ischaemia is clearly indicative that lymphatic drainage is insufficient for fluid drainage. At early stages post-MI, histopathologic analysis of patients with acute MI reveals the progressive loss of lymphatic vessels from the interstitium, as compared with the normal myocardium. Nevertheless, studies using mouse models of myocardial I/R and MI have shown that normal lymphatic density is subsequently restored at later stages post-MI; in the subendocardial compartment, lymphatic density was significantly augmented 3 days post-MI and gradually increased until day 7 [69]. This augmentation is likely to be driven by VEGF-C, the main lymphangiogenic growth factor acting through VEGFR3 during embryonic development. VEGF-C is expressed in the cardiomyocytes around the cardiac lesion and as such may act as a source for the restoration of cardiac lymphatic vessels [100]. Additionally, macrophages and the epicardium represent another source of VEGF-C post injury [74][101] and neutrophils have been described as organisers of lymphangiogenesis during inflammation by increasing VEGF-A bioavailability and secreting VEGF-D [102]. However, in rats, despite the significant increase in the density of lymphatic capillaries at 4 weeks post-MI, the percentages of pre-collector vessels, as well as open lymphatics and their diameters were decreased, likely explaining the poor lymphatic draining capacity and the subsequent persistence of myocardial oedema [66]. The death of cardiomyocytes during MI reduces cardiac contractility, which in turn impedes lymph propulsion from the heart to MLNs [103], and may also be responsible for inefficient fluid drainage.
As discussed above, sub-optimal heart recovery following MI is mainly due to the persistence of immune cells in the infarcted zone that delay or prevent the resolution of inflammation and the timely repair of cardiac injury. Given that one of the primary functions of lymphatic vessels is immune cell clearance from injured tissue, several groups have investigated the importance of lymphangiogenesis post-MI. Post-MI treatment with VEGF-C(C156S), an artificially mutated form of VEGF-C that binds exclusively to VEGFR3, induces a lymphangiogenic response in the rodent heart that results in an improvement of cardiac function [56][66][69][74][104]. In a mouse model of MI, intraperitoneal (i.p.) injection of VEGF-C(C156S) augmented cardiac lymphangiogenesis after injury. Interestingly, in the treated group the numbers of infiltrating leukocytes, including macrophages and DCs were shown to be significantly reduced 7 days post-MI, indicating enhanced clearance of immune cells to the MLNs. On the contrary, LYVE-1 gene deletion was shown to worsen cardiac outcomes and to promote chronic inflammation, due to the reduced ability of Lyve1−/− lymphatics to clear the immune cells [74]. Curiously, Houssari et al., using the same approach, failed to observe a significant increase of lymphangiogenesis after i.p injection of VEGF-C(C156S) [104]. However, it is important to note that these workers quantified cardiac lymphatic vessel density by conventional histology, whereas the former study used whole-mount LYVE1 immunostaining ([104] and [74] respectively) which could explain the discrepant findings. Moreover, cardiac function and heart remodelling post-MI after VEGF-C(C156S) treatment were not evaluated in the Houssari study, making a conclusion based exclusively on standard histology difficult to reconcile. Nevertheless, they also employed a different approach with an i.p injection of an adeno-associated viral vector encoding VEGF-C(C156S) (AAV-VEGF-C(C156S)) 7 days before MI and observed an increase in lymphangiogenesis 7 days post-MI as well as a decrease in both T-cells and pro-inflammatory macrophages in the viable left ventricle but not in the infarcted area 21 days post-MI. Fractional shortening was increased in mice treated with AAV-VEGF-C(C156S) therapy, indicating an improvement of cardiac function [104]. Another group adopted an intramyocardial, targeted delivery of VEGF-C(C156S) using albumin-alginate microparticles in a rat model. Here, high doses of the growth factor significantly increased the lymphatic density in the subepicardium by 3 weeks post-MI. 8 weeks post-MI, the frequency of larger epicardial pre-collectors was increased in treated rats and contributed to improved cardiac lymphatic drainage. 3 weeks post-MI, myocardial water balance was also improved, and the numbers of macrophages in the infarcted left ventricle were reduced significantly. MRI and echocardiography analysis confirmed the therapeutic lymphangiogenic effect on cardiac perfusion and function [66]. To further investigate this, another team used hydrogel as a strategy for VEGF-C(C156S) delivery. In a mouse myocardial I/R model, the gel was placed on the surface of the myocardium at the time of re-perfusion. Seven days later, the lymphatic density increased, and the number of B-cells decreased, as did myocardial oedema and the levels of various pro-inflammatory cytokines such as TNF-α, IL1β, and IL-6. 28 days post-reperfusion, the infarct scar, the LV end-diastolic diameter, and LV end-systolic diameter were all reduced, while the ejection fraction was improved in comparison with control mice, showing that hydrogel containing VEGF-C(C156S) can indeed limit heart failure. Conversely, inhibiting VEGFR3 or VEGF-C with a neutralising antibody exerted the opposite effect and aggravated cardiac dysfunction [69]. Thus, collectively there is now substantial evidence that delivery of VEGF-C(C156S) by a variety of routes in experimental animal models can significantly improve the outcome post-MI; through targeting increased lymphangiogenesis to reduce oedema and enhance the clearance of immune cells.
The half-life of VEGF-C is extremely short [105] which suggests it is not an ideal target for therapeutic use. Clinical trials using intramyocardial adenovirus vector-mediated VEGFD-ΔNΔC gene therapy in patients with refractory angina have established the safety and feasibility of this therapy, accompanied by a positive outcome in treated patients with an increase of the myocardial perfusion [106]. Although quite promising, the approach nevertheless remains invasive, with the necessity for repeated injections in the myocardium that have a high cost per patient. Hence, it will be necessary to find other means of manipulating lymphangiogenesis (growth factors, compounds, existing drugs) to gain a better understanding of the mechanisms involved, and to discover potential new treatments for heart repair.
One alternative strategy, overexpression of the epicardium-specific peptide, adrenomedullin, has recently been shown to trigger lymphangiogenesis post-MI and to improve cardiac function in mice. Interestingly, sex-dependent differences were noted, with a decrease in myocardial oedema that was found exclusively in males. Furthermore, ejection fraction and fractional shortening were improved after only 10 days in females versus 15 days in males. This indicates an important limiting factor in the discovery of novel lymphangiogenic compounds, as in the steady-state heart, cardiac lymphatic density also differs between males and females [107]. The chemoattractant Shingosine-1-phosphate (S1P) has been described as another lymphangiogenic mediator both in vitro and in vivo [108] and may have a role in lymphatic vessel maturation [109]. In addition, S1P has been well described as a lipid mediator of leucocyte egress from lymphoid organs [110]. Though it has also been implicated in DC trafficking [111], its role in the trafficking of immune cells from inflamed tissues to dLNs is poorly understood and warrants further investigations especially in the context of MI.
Finally, studies have also focused on cell-based therapies to increase lymphangiogenesis and restore heart function post-MI. Using a rat model of MI, Zhang et al. investigated the potential effects of transplanting lymphatic endothelial cell progenitors (LECP) either alone or in combination with VEGF-C, using a self-assembling peptide (SAP) hydrogel that facilitated a sustained release of the growth factor [112][113]. Individually, both treatment strategies led to an improvement in cardiac function and their combination yielded an additive effect by significantly reducing myocardial oedema and fibrotic scar size. Moreover, the numbers of infiltrating immune cells correlated inversely with the number of lymphatic vessels and both the ejection fraction, and the fractional shortening were restored in the treated rats [112]. Hence, this represents a feasible strategy for therapeutic use in the future. Cardiac fibroblasts are another cell type with potential beneficial properties for heart repair. In particular, a specific subpopulation expressing VCAM-1 (CFV) has been identified as a potential inducer of lymphangiogenesis. This population expresses several pro-lymphangiogenic factors, including VEGF-C, and was shown to promote lymphangiogenesis as assessed by assays for in vitro tube formation. Furthermore, injection of human foetal CFV in post-infarct heart failure rat models mobilised LECs into the infarcted area and restored cardiac contractility [114].
In addition to clearing immune cells and resolving myocardial oedema, it has been recently shown that cardiac lymphatics may confer other beneficial effects on heart recovery post-MI. LECs secrete a variety of growth factors, cytokines, and chemokines known as “lymphangiocrine” factors, that are active during the initiation of immune responses [115]. The LEC secretome contains the extracellular protein reelin which promotes cardiomyocyte proliferation and survival. Cardiac delivery of reelin post-MI, in mice, improves heart function by exerting a cardioprotective effect [116].
Additionally, lymphangiogenic therapy also has a positive outcome in other cardiac diseases. In human chronic heart failure, the levels of lymphatic endothelial markers are decreased in comparison with healthy donors [117]. Using the Ang2 infusion-induced mouse model, Song et al. demonstrated that co-administration of VEGF-C(C156S) prevented cardiac dysfunction due to an improvement of the cardiac lymphatic vascular function and a decrease of the inflammatory response. After one week, inflammatory and fibrosis markers were decreased, and after five weeks, hypertension in the VEGF-C(C156S) treated group was almost abolished [117]. This is important, since survival from acute injury event in MI patients is significant, however, subsequent progression to heart failure arising from pathological remodelling remains a major cause of morbidity and mortality for which the only cure is heart transplantation. Furthermore, advances in 3D imaging of the cardiac lymphatic vasculature in vivo in mice has identified that increasing lymphatic vascular density may not be enough to resolve inflammation and cardiac oedema [118]. Furthermore, advances in imaging in humans have also highlighted the importance of addressing lymph flow when addressing heart disease [119][120]. Therefore, ensuring there is increased lymph flow, in addition to enhanced lymphangiogenesis, is also critical in the trafficking of immune cells from the site of injury and may also be a potential therapeutic approach [118][119]. Targeting the lymphatics during the chronic phase post-MI, therefore, to potentially alleviate the major drivers of heart failure presents an attractive target for therapeutic lymphangiogenesis.
References
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell Cardiol. 2013, 62, 24–35.
- Pittman, K.; Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 2013, 5, 315–323.
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol 2011, 8, 292–300.
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61.
- Prince, L.R.; Whyte, M.K.; Sabroe, I.; Parker, L.C. The role of TLRs in neutrophil activation. Curr. Opin. Pharmacol. 2011, 11, 397–403.
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 2021, 77, 109816.
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197.
- Chia, S.; Nagurney, J.T.; Brown, D.F.; Raffel, O.C.; Bamberg, F.; Senatore, F.; Wackers, F.J.; Jang, I.K. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2009, 103, 333–337.
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127.
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104.
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551.
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295.
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511.
- Cahill, T.J.; Sun, X.; Ravaud, C.; Villa Del Campo, C.; Klaourakis, K.; Lupu, I.E.; Lord, A.M.; Browne, C.; Jacobsen, S.E.W.; Greaves, D.R.; et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development 2021, 148.
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28.
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278.
- Li, W.; Hsiao, H.M.; Higashikubo, R.; Saunders, B.T.; Bharat, A.; Goldstein, D.R.; Krupnick, A.S.; Gelman, A.E.; Lavine, K.J.; Kreisel, D. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016, 1.
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39.
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889.
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047.
- Elkenhans, B.; Protti, A.; Shah, A.; Onthank, D.; Botnar, R. Visualization of elastin using cardiac magnetic resonance imaging after myocardial infarction as inflammatory response. Sci. Rep. 2021, 11, 1–9.
- Frangogiannis, N.G.; Mendoza, L.H.; Ren, G.; Akrivakis, S.; Jackson, P.L.; Michael, L.H.; Smith, C.W.; Entman, M.L. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H483–H492.
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112.
- Molenaar, B.; Timmer, L.T.; Droog, M.; Perini, I.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; de Ruiter, H.; Gladka, M.M.; van Rooij, E. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 2021, 4, 1–15.
- O’Rourke, S.A.; Dunne, A.; Monaghan, M.G. The Role of Macrophages in the Infarcted Myocardium: Orchestrators of ECM Remodeling. Front. Cardiovasc. Med. 2019, 6, 101.
- Van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 2007, 170, 818–829.
- Frangogiannis, N.G.; Ren, G.; Dewald, O.; Zymek, P.; Haudek, S.; Koerting, A.; Winkelmann, K.; Michael, L.H.; Lawler, J.; Entman, M.L. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 2005, 111, 2935–2942.
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428.
- Ma, Y.; de Castro Bras, L.E.; Toba, H.; Iyer, R.P.; Hall, M.E.; Winniford, M.D.; Lange, R.A.; Tyagi, S.C.; Lindsey, M.L. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflug. Arch. 2014, 466, 1113–1127.
- Simoes, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600.
- Satoh, T.; Kidoya, H.; Naito, H.; Yamamoto, M.; Takemura, N.; Nakagawa, K.; Yoshioka, Y.; Morii, E.; Takakura, N.; Takeuchi, O.; et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 2013, 495, 524–528.
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Invest. 2016, 126, 2151–2166.
- Al-Darraji, A.; Haydar, D.; Chelvarajan, L.; Tripathi, H.; Levitan, B.; Gao, E.; Venditto, V.J.; Gensel, J.C.; Feola, D.J.; Abdel-Latif, A. Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: Potential therapeutic targets in ischemic heart disease. PLoS ONE 2018, 13, e0200474.
- Shintani, Y.; Ito, T.; Fields, L.; Shiraishi, M.; Ichihara, Y.; Sato, N.; Podaru, M.; Kainuma, S.; Tanaka, H.; Suzuki, K. IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice. Sci. Rep. 2017, 7, 1–7.
- Peng, Y.; Chen, B.; Zhao, J.; Peng, Z.; Xu, W.; Yu, G. Effect of intravenous transplantation of hUCB-MSCs on M1/M2 subtype conversion in monocyte/macrophages of AMI mice. Biomed. Pharmacother. 2019, 111, 624–630.
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564.
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715.
- Dai, H.; Thomson, A.W.; Rogers, N.M. Dendritic Cells as Sensors, Mediators, and Regulators of Ischemic Injury. Front. Immunol. 2019, 10, 2418.
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouche, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017.
- Forte, E.; Perkins, B.; Sintou, A.; Kalkat, H.S.; Papanikolaou, A.; Jenkins, C.; Alsubaie, M.; Chowdhury, R.A.; Duffy, T.M.; Skelly, D.A.; et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation 2021, 143, 821–836.
- Satpathy, A.T.; Kc, W.; Albring, J.C.; Edelson, B.T.; Kretzer, N.M.; Bhattacharya, D.; Murphy, T.L.; Murphy, K.M. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 2012, 209, 1135–1152.
- Lee, J.S.; Jeong, S.J.; Kim, S.; Chalifour, L.; Yun, T.J.; Miah, M.A.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798.
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Gunther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834.
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245.
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.A.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 2014, 3, e000839.
- Choo, E.H.; Lee, J.H.; Park, E.H.; Park, H.E.; Jung, N.C.; Kim, T.H.; Koh, Y.S.; Kim, E.; Seung, K.B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation 2017, 135, 1444–1457.
- Zhu, R.; Sun, H.; Yu, K.; Zhong, Y.; Shi, H.; Wei, Y.; Su, X.; Xu, W.; Luo, Q.; Zhang, F.; et al. Interleukin-37 and Dendritic Cells Treated With Interleukin-37 Plus Troponin I Ameliorate Cardiac Remodeling After Myocardial Infarction. J. Am. Heart Assoc. 2016, 5.
- Klaourakis, K.; Vieira, J.M.; Riley, P.R. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat. Rev. Cardiol. 2021, 18, 368–379.
- Park, C.; Kim, T.M.; Malik, A.B. Transcriptional regulation of endothelial cell and vascular development. Circ. Res. 2013, 112, 1380–1400.
- Wigle, J.T.; Oliver, G. Prox1 function is required for the development of the murine lymphatic system. Cell 1999, 98, 769–778.
- Gale, N.W.; Thurston, G.; Hackett, S.F.; Renard, R.; Wang, Q.; McClain, J.; Martin, C.; Witte, C.; Witte, M.H.; Jackson, D.; et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 2002, 3, 411–423.
- Harvey, N.L.; Srinivasan, R.S.; Dillard, M.E.; Johnson, N.C.; Witte, M.H.; Boyd, K.; Sleeman, M.W.; Oliver, G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat. Genet. 2005, 37, 1072–1081.
- Hong, Y.K.; Harvey, N.; Noh, Y.H.; Schacht, V.; Hirakawa, S.; Detmar, M.; Oliver, G. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev. Dyn. 2002, 225, 351–357.
- Kim, H.; Nguyen, V.P.; Petrova, T.V.; Cruz, M.; Alitalo, K.; Dumont, D.J. Embryonic vascular endothelial cells are malleable to reprogramming via Prox1 to a lymphatic gene signature. BMC Dev. Biol. 2010, 10, 1–10.
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2003, 5, 74–80.
- Klotz, L.; Norman, S.; Vieira, J.M.; Masters, M.; Rohling, M.; Dube, K.N.; Bollini, S.; Matsuzaki, F.; Carr, C.A.; Riley, P.R. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 2015, 522, 62–67.
- Ulvmar, M.H.; Martinez-Corral, I.; Stanczuk, L.; Makinen, T. Pdgfrb-Cre targets lymphatic endothelial cells of both venous and non-venous origins. Genesis 2016, 54, 350–358.
- Makinen, T.; Jussila, L.; Veikkola, T.; Karpanen, T.; Kettunen, M.I.; Pulkkanen, K.J.; Kauppinen, R.; Jackson, D.G.; Kubo, H.; Nishikawa, S.; et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat. Med. 2001, 7, 199–205.
- Chen, H.I.; Sharma, B.; Akerberg, B.N.; Numi, H.J.; Kivela, R.; Saharinen, P.; Aghajanian, H.; McKay, A.S.; Bogard, P.E.; Chang, A.H.; et al. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 2014, 141, 4500–4512.
- Juszynski, M.; Ciszek, B.; Stachurska, E.; Jablonska, A.; Ratajska, A. Development of lymphatic vessels in mouse embryonic and early postnatal hearts. Dev. Dyn. 2008, 237, 2973–2986.
- Bradham, R.R.; Parker, E.F. The cardiac lymphatics. Ann. Thorac. Surg. 1973, 15, 526–535.
- Flaht-Zabost, A.; Gula, G.; Ciszek, B.; Czarnowska, E.; Jankowska-Steifer, E.; Madej, M.; Niderla-Bielińska, J.; Radomska-Leśniewska, D.; Ratajska, A. Cardiac Mouse Lymphatics: Developmental and Anatomical Update. Anat. Rec. 2014, 297, 1115–1130.
- Ratajska, A.; Gula, G.; Flaht-Zabost, A.; Czarnowska, E.; Ciszek, B.; Jankowska-Steifer, E.; Niderla-Bielinska, J.; Radomska-Lesniewska, D. Comparative and Developmental Anatomy of Cardiac Lymphatics. Sci. World J. 2014, 2014.
- Shore, L.R. The Lymphatic Drainage of the Human Heart. J. Anat. 1929, 63, 291–313.
- Shimada, T.; Zhang, L.; Abe, K.; Yamabe, M.; Miyamoto, T. Developmental morphology of blood and lymphatic capillary networks in mammalian hearts, with special reference to three-dimensional architecture. Ital. J. Anat. Embryol. 2001, 106, 203–211.
- Henri, O.; Pouehe, C.; Houssari, M.; Galas, L.; Nicol, L.; Edwards-Levy, F.; Henry, J.P.; Dumesnil, A.; Boukhalfa, I.; Banquet, S.; et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 2016, 133, 1484–1497.
- Geissler, H.; Allen, S.; Davis, K.; Sauer, H.; Laine, G.; Kuhn-Régnier, F.; Dapunt, O.; Vivie, E.d.; Mehlhorn, U. Impact of cardiopulmonary bypass and cardioplegic arrest on myocardial efficiency. Crit. Care 1999, 3, 1–2.
- Laine, G.A.; Allen, S.J. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ. Res. 1991, 68, 1713–1721.
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Pantner, Y.; Nicholson, C.K.; Ishii, M.; Brunnhoelzl, D.; Mauria, R.; Husain, A.; Naqvi, N.; et al. Impact of Lymphangiogenesis on Cardiac Remodeling After Ischemia and Reperfusion Injury. J. Am. Heart Assoc. 2018, 7, e009565.
- Kholova, I.; Dragneva, G.; Cermakova, P.; Laidinen, S.; Kaskenpaa, N.; Hazes, T.; Cermakova, E.; Steiner, I.; Yla-Herttuala, S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur. J. Clin. Invest. 2011, 41, 487–497.
- Geissler, H.J.; Dashkevich, A.; Fischer, U.M.; Fries, J.W.; Kuhn-Regnier, F.; Addicks, K.; Mehlhorn, U.; Bloch, W. First year changes of myocardial lymphatic endothelial markers in heart transplant recipients. Eur. J. Cardiothorac. Surg. 2006, 29, 767–771.
- Rutanen, J.; Leppanen, P.; Tuomisto, T.T.; Rissanen, T.T.; Hiltunen, M.O.; Vajanto, I.; Niemi, M.; Hakkinen, T.; Karkola, K.; Stacker, S.A.; et al. Vascular endothelial growth factor-D expression in human atherosclerotic lesions. Cardiovasc. Res. 2003, 59, 971–979.
- Dongaonkar, R.M.; Stewart, R.H.; Geissler, H.J.; Laine, G.A. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc. Res. 2010, 87, 331–339.
- Vieira, J.M.; Norman, S.; Villa Del Campo, C.; Cahill, T.J.; Barnette, D.N.; Gunadasa-Rohling, M.; Johnson, L.A.; Greaves, D.R.; Carr, C.A.; Jackson, D.G.; et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest. 2018, 128, 3402–3412.
- Frantz, S.; Hofmann, U.; Fraccarollo, D.; Schafer, A.; Kranepuhl, S.; Hagedorn, I.; Nieswandt, B.; Nahrendorf, M.; Wagner, H.; Bayer, B.; et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013, 27, 871–881.
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285.
- Jantsch, J.; Binger, K.J.; Muller, D.N.; Titze, J. Macrophages in homeostatic immune function. Front. Physiol. 2014, 5, 146.
- Kastenmuller, W.; Torabi-Parizi, P.; Subramanian, N.; Lammermann, T.; Germain, R.N. A spatially organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 2012, 150, 1235–1248.
- Von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol 2003, 3, 867–878.
- Bellingan, G.J.; Caldwell, H.; Howie, S.E.; Dransfield, I.; Haslett, C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: Inflammatory macrophages do not die locally but emigrate to the draining lymph nodes. J. Immunol. 1996, 157, 2577–2585.
- Hou, Y.; Bock, F.; Hos, D.; Cursiefen, C. Lymphatic Trafficking in the Eye: Modulation of Lymphatic Trafficking to Promote Corneal Transplant Survival. Cells 2021, 10, 1661.
- Bellingan, G.J.; Xu, P.; Cooksley, H.; Cauldwell, H.; Shock, A.; Bottoms, S.; Haslett, C.; Mutsaers, S.E.; Laurent, G.J. Adhesion molecule-dependent mechanisms regulate the rate of macrophage clearance during the resolution of peritoneal inflammation. J. Exp. Med. 2002, 196, 1515–1521.
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature- Mechanisms and Consequences. Front. Immunol. 2019, 10, 471.
- Huang, F.P.; Platt, N.; Wykes, M.; Major, J.R.; Powell, T.J.; Jenkins, C.D.; MacPherson, G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000, 191, 435–444.
- Jakubzick, C.; Bogunovic, M.; Bonito, A.J.; Kuan, E.L.; Merad, M.; Randolph, G.J. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J. Exp. Med. 2008, 205, 2839–2850.
- MacPherson, G.G.; Jenkins, C.D.; Stein, M.J.; Edwards, C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J. Immunol. 1995, 154, 1317–1322.
- Gorlino, C.V.; Ranocchia, R.P.; Harman, M.F.; Garcia, I.A.; Crespo, M.I.; Moron, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J. Immunol. 2014, 193, 1966–1974.
- Chtanova, T.; Schaeffer, M.; Han, S.J.; van Dooren, G.G.; Nollmann, M.; Herzmark, P.; Chan, S.W.; Satija, H.; Camfield, K.; Aaron, H.; et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity 2008, 29, 487–496.
- Beauvillain, C.; Cunin, P.; Doni, A.; Scotet, M.; Jaillon, S.; Loiry, M.L.; Magistrelli, G.; Masternak, K.; Chevailler, A.; Delneste, Y.; et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood 2011, 117, 1196–1204.
- Forster, R.; Braun, A.; Worbs, T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012, 33, 271–280.
- Daroczy, J. The structure and dynamic function of the dermal lymphatic capillaries. Br. J. Dermatol. 1983, 109, 99–102.
- Schmid-Schonbein, G.W. The second valve system in lymphatics. Lymphat. Res. Biol. 2003, 1, 25–31.
- Bazigou, E.; Wilson, J.T.; Moore, J.E., Jr. Primary and secondary lymphatic valve development: Molecular, functional and mechanical insights. Microvasc. Res. 2014, 96, 38–45.
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575.
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362.
- Romani, N.; Ratzinger, G.; Pfaller, K.; Salvenmoser, W.; Stossel, H.; Koch, F.; Stoitzner, P. Migration of dendritic cells into lymphatics-the Langerhans cell example: Routes, regulation, and relevance. Int. Rev. Cytol. 2001, 207, 237–270.
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935.
- Scallan, J.P.; Zawieja, S.D.; Castorena-Gonzalez, J.A.; Davis, M.J. Lymphatic pumping: Mechanics, mechanisms and malfunction. J. Physiol. 2016, 594, 5749–5768.
- Tatin, F.; Renaud-Gabardos, E.; Godet, A.C.; Hantelys, F.; Pujol, F.; Morfoisse, F.; Calise, D.; Viars, F.; Valet, P.; Masri, B.; et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight 2017, 2.
- Ishikawa, Y.; Akishima-Fukasawa, Y.; Ito, K.; Akasaka, Y.; Tanaka, M.; Shimokawa, R.; Kimura-Matsumoto, M.; Morita, H.; Sato, S.; Kamata, I.; et al. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology 2007, 51, 345–353.
- Kataru, R.P.; Jung, K.; Jang, C.; Yang, H.; Schwendener, R.A.; Baik, J.E.; Han, S.H.; Alitalo, K.; Koh, G.Y. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood 2009, 113, 5650–5659.
- Tan, K.W.; Chong, S.Z.; Wong, F.H.; Evrard, M.; Tan, S.M.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677.
- Taira, A.; Morishita, Y.; Arikawa, K.; Murata, K.; Hamada, Y.; Akita, H. Flow velocity of cardiac lymph and contractility of the heart: An experimental study. Ann. Thorac. Surg. 1977, 23, 230–234.
- Houssari, M.; Dumesnil, A.; Tardif, V.; Kivela, R.; Pizzinat, N.; Boukhalfa, I.; Godefroy, D.; Schapman, D.; Hemanthakumar, K.A.; Bizou, M.; et al. Lymphatic and Immune Cell Cross-Talk Regulates Cardiac Recovery After Experimental Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2020, 40, 1722–1737.
- Veikkola, T.; Jussila, L.; Makinen, T.; Karpanen, T.; Jeltsch, M.; Petrova, T.V.; Kubo, H.; Thurston, G.; McDonald, D.M.; Achen, M.G.; et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001, 20, 1223–1231.
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivela, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DDeltaNDeltaC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555.
- Trincot, C.E.; Xu, W.; Zhang, H.; Kulikauskas, M.R.; Caranasos, T.G.; Jensen, B.C.; Sabine, A.; Petrova, T.V.; Caron, K.M. Adrenomedullin Induces Cardiac Lymphangiogenesis After Myocardial Infarction and Regulates Cardiac Edema Via Connexin 43. Circ. Res. 2019, 124, 101–113.
- Yoon, C.M.; Hong, B.S.; Moon, H.G.; Lim, S.; Suh, P.G.; Kim, Y.K.; Chae, C.B.; Gho, Y.S. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood 2008, 112, 1129–1138.
- Pham, T.H.; Baluk, P.; Xu, Y.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27.
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94.
- Czeloth, N.; Bernhardt, G.; Hofmann, F.; Genth, H.; Forster, R. Sphingosine-1-phosphate mediates migration of mature dendritic cells. J. Immunol. 2005, 175, 2960–2967.
- Zhang, H.F.; Wang, Y.L.; Tan, Y.Z.; Wang, H.J.; Tao, P.; Zhou, P. Enhancement of cardiac lymphangiogenesis by transplantation of CD34(+)VEGFR-3(+) endothelial progenitor cells and sustained release of VEGF-C. Basic Res. Cardiol. 2019, 114, 1–17.
- Boopathy, A.V.; Davis, M.E. Self-assembling peptide-based delivery of therapeutics for myocardial infarction. Methods Mol. Biol. 2014, 1141, 159–164.
- Iwamiya, T.; Segard, B.D.; Matsuoka, Y.; Imamura, T. Human cardiac fibroblasts expressing VCAM1 improve heart function in postinfarct heart failure rat models by stimulating lymphangiogenesis. PLoS ONE 2020, 15, e0237810.
- Lee, E.; Pandey, N.B.; Popel, A.S. Lymphatic endothelial cells support tumor growth in breast cancer. Sci. Rep. 2014, 4, 5853.
- Liu, X.; De la Cruz, E.; Gu, X.; Balint, L.; Oxendine-Burns, M.; Terrones, T.; Ma, W.; Kuo, H.H.; Lantz, C.; Bansal, T.; et al. Lymphoangiocrine signals promote cardiac growth and repair. Nature 2020, 588, 705–711.
- Song, L.; Chen, X.; Swanson, T.A.; LaViolette, B.; Pang, J.; Cunio, T.; Nagle, M.W.; Asano, S.; Hales, K.; Shipstone, A.; et al. Lymphangiogenic therapy prevents cardiac dysfunction by ameliorating inflammation and hypertension. Elife 2020, 9.
- Yeo, K.P.; Lim, H.Y.; Thiam, C.H.; Azhar, S.H.; Tan, C.; Tang, Y.; See, W.Q.; Koh, X.H.; Zhao, M.H.; Phua, M.L.; et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci. Adv. 2020, 6.
- Itkin, M.; Rockson, S.G.; Burkhoff, D. Pathophysiology of the Lymphatic System in Patients with Heart Failure: JACC State-of-the-Art Review. J. Am. Coll Cardiol. 2021, 78, 278–290.
- Sinha, S.; Lee, E.W.; Dori, Y.; Katsuhide, M. Advances in lymphatic imaging and interventions in patients with congenital heart disease. Prog. Pediatr. Cardiol. 2021, 61, 101376.
More
Information
Subjects:
Developmental Biology; Immunology; Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
705
Revisions:
2 times
(View History)
Update Date:
13 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No