 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ramon Cacabelos | -- | 2944 | 2022-04-12 12:25:23 | | | |
| 2 | Amina Yu | -34 word(s) | 2910 | 2022-04-13 04:02:48 | | |
Video Upload Options
Alzheimer’s disease (AD) is a priority health problem with a high cost to society and a large consumption of medical and social resources. The management of AD patients is complex and multidisciplinary. Over 90% of patients suffer from concomitant diseases and require personalized therapeutic regimens to reduce adverse drug reactions (ADRs), drug–drug interactions (DDIs), and unnecessary costs. Men and women show substantial differences in their AD-related phenotypes. Genomic, epigenetic, neuroimaging, and biochemical biomarkers are useful for predictive and differential diagnosis. The most frequent concomitant diseases include hypertension (>25%), obesity (>70%), diabetes mellitus type 2 (>25%), hypercholesterolemia (40%), hypertriglyceridemia (20%), metabolic syndrome (20%), hepatobiliary disorder (15%), endocrine/metabolic disorders (>20%), cardiovascular disorder (40%), cerebrovascular disorder (60–90%), neuropsychiatric disorders (60–90%), and cancer (10%). Over 90% of AD patients require multifactorial treatments with risk of ADRs and DDIs. The implementation of pharmacogenetics in clinical practice can help optimize the limited therapeutic resources available to treat AD and personalize the use of anti-dementia drugs, in combination with other medications, for the treatment of concomitant disorders.
1. Phenotypic Features
2. Biomarkers
2.1. Genomic Markers
2.2. Epigenetic Markers
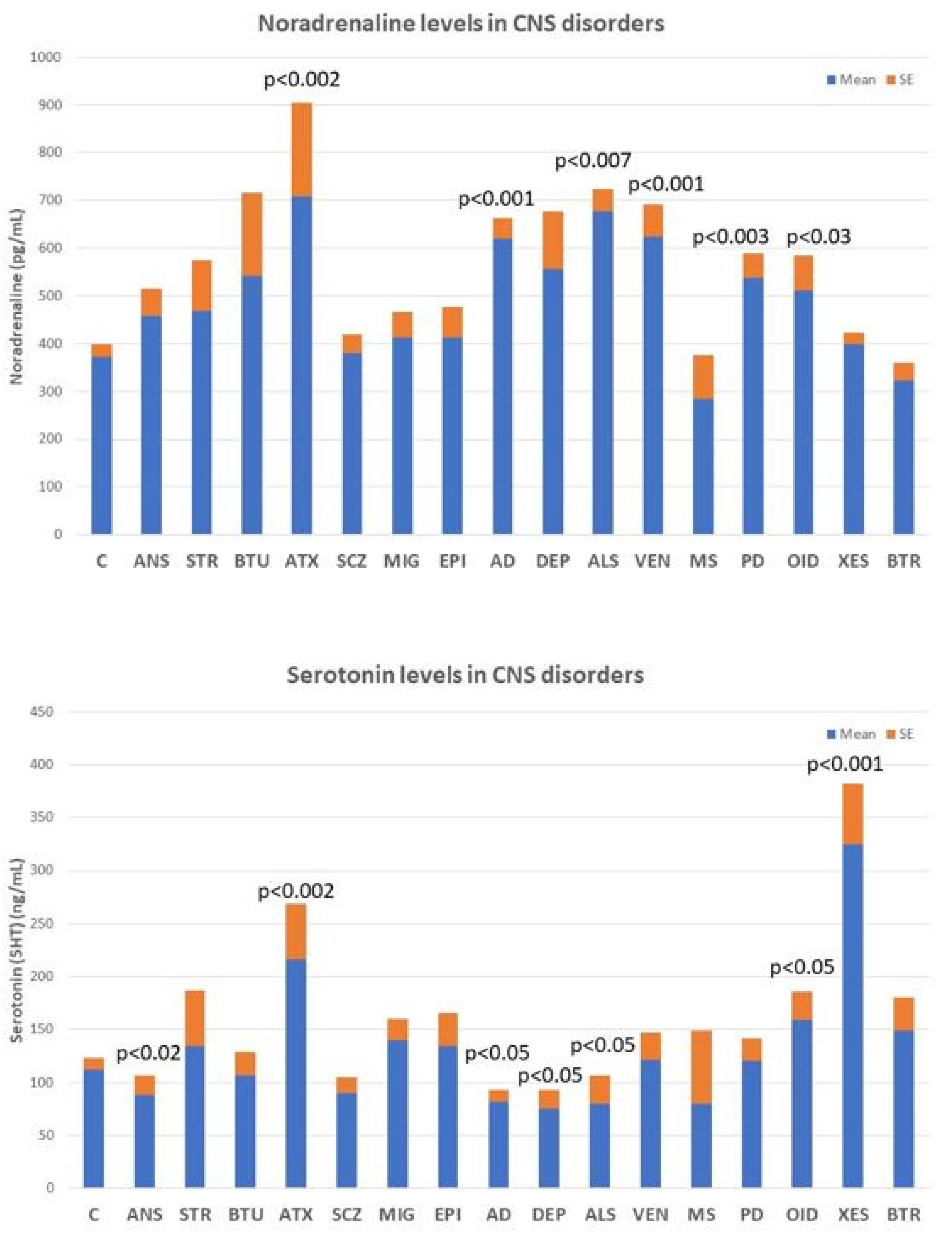
2.3. Neurotransmitters
2.4. Aβ/Tau Levels
3. Concomitant Disorders and Phenotype-Modifying Treatments
4. Alzheimer’s Disease Therapeutics and Drug Development
4.1. Immunotherapy
References
- Bature, F.; Guinn, B.A.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746.
- Linnemann, C.; Lang, U.E. Pathways Connecting Late-Life Depression and Dementia. Front. Pharmacol. 2020, 11, 279.
- Cacabelos, R.; Carril, J.C.; Corzo, L.; Fernández-Novoa, L.; Pego, R.; Cacabelos, N.; Cacabelos, P.; Alcaraz, M.; Tellado, I.; Naidoo, V. Influence of pathogenic and metabolic genes on the pharmacogenetics of mood disorders in Alzheimer’s disease. Pharmaceuticals 2021, 14, 366.
- Bennett, S.; Thomas, A.J. Depression and dementia: Cause, consequence or coincidence? Maturitas 2014, 79, 184–190.
- Gutzmann, H.; Qazi, A. Depression associated with dementia. Z. Gerontol. Geriatr. 2015, 48, 305–311.
- Baruch, N.; Burgess, J.; Pillai, M.; Allan, C.L. Treatment for depression comorbid with dementia. Evid. Based Ment. Health 2019, 22, 167–171.
- Bingham, K.S.; Flint, A.J.; Mulsant, B.H. Management of Late-Life Depression in the Context of Cognitive Impairment: A Review of the Recent Literature. Curr. Psychiatry Rep. 2019, 21, 74.
- Kratz, T. The Diagnosis and Treatment of Behavioral Disorders in Dementia. Dtsch. Arztebl. Int. 2017, 114, 447–454.
- Aarsland, D. Epidemiology and Pathophysiology of Dementia-Related Psychosis. J. Clin. Psychiatry 2020, 81, AD19038BR1C.
- Cacabelos, R. Pharmacogenomic Biomarkers in Neuropsychiatry: The Path to Personalized Medicine in Mental Disorders. In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Molecular Genetic and Genomic Markers; Ritsner, M.S., Ed.; Springer: Amsterdam, The Netherlands, 2009; Volume 4, pp. 3–63.
- Caroli, A.; Frisoni, G.B.; Alzheimer’s Disease Neuroimaging Initiative. The dynamics of Alzheimer’s disease biomarkers in the Alzheimer’s Disease Neuroimaging Initiative cohort. Neurobiol. Aging 2010, 31, 1263–1274.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Peterson, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Wigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216.
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663.
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s disease: Preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2021, 26, 296–308.
- Márquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 21.
- Cacabelos, R. Genomic characterization of Alzheimer’s disease and genotype-related phenotypic analysis of biological markers in dementia. Pharmacogenomics 2004, 5, 1049–1105.
- Cacabelos, R.; Fernández-Novoa, L.; Corzo, L.; Amado, L.; Pichel, V.; Lombardi, V.; Kubota, Y. Phenotypic profiles and functional genomics in Alzheimer’s disease and in dementia with a vascular component. Neurol. Res. 2004, 26, 459–480.
- Cacabelos, R.; Fernández-Novoa, L.; Corzo, L.; Pichel, V.; Lombardi, V.; Kubota, Y. Genomics and phenotypic profiles in dementia: Implications for pharmacological treatment. Methods Find. Exp. Clin. Pharmacol. 2004, 26, 421–444.
- Cacabelos, R.; Lombardi, V.; Fernández-Novoa, L.; Kubota, Y.; Corzo, L.; Pichel, V.; Takeda, M. A functional genomics approach to the analysis of biological markers in Alzheimer disease. In Molecular Neurobiology of Alzheimer’s Disease and Related Disorders; Takeda, M., Tanaka, T., Cacabelos, R., Eds.; Karger: Basel, Switzerland, 2004; pp. 236–285.
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 1–573.
- Cacabelos, R.; Martínez-Bouza, R.; Carril, J.C.; Fernández-Novoa, L.; Lombardi, V.; Carrera, I.; Corzo, L.; McKay, A. Genomics and pharmacogenomics of brain disorders. Curr. Pharm. Biotechnol. 2012, 13, 674–725.
- Cacabelos, R.; Martínez-Bouza, R. Genomics and pharmacogenomics of dementia. CNS Neurosc. Ther. 2011, 17, 566–576.
- Cai, Y.; An, S.S.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172.
- Kelleher, R.J.; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631.
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560.
- Giau, V.V.; Bagyinszky, E.; Yang, Y.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368.
- Van Cauwenberghe, C.; van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430.
- Bertram, L. Next Generation Sequencing in Alzheimer’s Disease. Syst. Biol. Alzheimers Dis. 2016, 1303, 281–297.
- Zhu, J.B.; Tan, C.C.; Tan, L.; Yu, J.T. State of Play in Alzheimer’s Disease Genetics. J. Alzheimers Dis. 2017, 58, 631–659.
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51.
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556.
- Cacabelos, R.; Carril, J.C.; Cacabelos, P.; Teijido, O.; Goldgaber, D. Pharmacogenomics of Alzheimer’s Disease: Genetic determinants of phenotypic variation and therapeutic outcome. J. Genom. Med. Pharmacogenom. 2016, 1, 151–209.
- Cacabelos, R.; Carril, J.C.; Cacabelos, N.; Kazantsev, A.G.; Vostrov, A.V.; Corzo, L.; Cacabelos, P.; Goldgaber, D. Sirtuins in Alzheimer’s Disease: SIRT2-Related GenoPhenotypes and Implications for PharmacoEpiGenetics. Int. J. Mol. Sci. 2019, 20, 1249.
- Cacabelos, R.; Goldgaber, D.; Vostrov, A.; Matsuki, H.; Torrellas, C.; Corzo, L.; Carril, J.C.; Roses, A.D. APOE-TOMM40 in the Pharmacogenomics of demetia. J. Pharmacogen. Pharmacoproteom. 2014, 5, 135.
- Cacabelos, R.; Torrellas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s disease. Pharmacogenomics 2016, 17, 1041–1074.
- Cacabelos, R.; Meyyazhagan, A.; Carril, J.C.; Cacabelos, P.; Teijido, O. Pharmacogenetics of vascular risk factors in Alzheimer’s Disease. J. Pers. Med. 2018, 8, 3.
- Cacabelos, R. Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev. Res. 2012, 73, 461–476.
- Cacabelos, R.; Takeda, M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future 2006, 3, 5–146.
- Cacabelos, R. Psychogeriatric research: A conceptual introduction to aging and geriatric neuroscience. Psychogeriatrics 2001, 1, 158–188.
- Cacabelos, R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics 2003, 4, 597–621.
- Cacabelos, R. Pharmacogenomics in Alzheimer’s disease. Pharm. Drug Discov. Dev. 2008, 448, 213–357.
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100.
- Cacabelos, R. Molecular pathology and pharmacogenomics in Alzheimer’s disease: Polygenic-related effects of multifactorial treatments on cognition, anxiety, and depression. Methods Find. Expert Clin. Pharmacol. 2007, 29, 1–91.
- Sabbagh, M.N.; Malek-Ahmadi, M.; Dugger, B.N.; Lee, K.; Sue, L.I.; Serrano, G.; Walker, D.G.; Davis, K.; Jacobson, S.A.; Beach, T.G. The influence of Apolipoprotein E genotype on regional pathology in Alzheimer’s disease. BMC Neurol. 2013, 13, 44.
- Kennedy, R.E.; Cutter, G.R.; Schneider, L.S. Effect of APOE genotype status on targeted clinical trials outcomes and efficiency in dementia and mild cognitive impairment resulting from Alzheimer’s disease. Alzheimers Dement. 2014, 10, 349–359.
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667.
- Cacabelos, R.; Torrellas, C.; López-Muñoz, F. Epigenomics of Alzheimer’s disease. J. Exp. Med. 2014, 6, 75–82.
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086.
- Cacabelos, R. (Ed.) Pathoepigenetics: The role of epigenetic biomarkers in disease pathogenesis. In Pharmacoepigenetics, 1st ed.; Academic Press: Oxford, UK, 2019; pp. 139–189.
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044.
- Ciceri, F.; Rotllant, D.; Maes, T. Understanding Epigenetic Alterations in Alzheimer’s and Parkinson’s Disease: Towards Targeted Biomarkers and Therapies. Curr. Pharm. Des. 2017, 23, 839–857.
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167.
- Gupta, P.; Bhattacharjee, S.; Sharma, A.R.; Sharma, G.; Lee, S.S.; Chakraborty, C. miRNAs in Alzheimer Disease—A Therapeutic Perspective. Curr. Alzheimer Res. 2017, 14, 1198–1206.
- Silvestro, S.; Bramanti, P.; Mazzon, E. Role of miRNAs in Alzheimer’s Disease and Possible Fields of Application. Int. J. Mol. Sci. 2019, 20, 3979.
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26.
- Nunomura, A.; Perry, G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on microRNAs. Oxidative Med. Cell. Longev. 2020, 2020, 2638130.
- Cacabelos, R.; Carril, J.C.; Sanmartín, A.; Cacabelos, P. Pharmacoepigenetic processors: Epigenetic drugs, Drug resistance, Toxicoepigenetics, and Nutriepigenetics. In Pharmacoepigenetics, 1st ed.; Cacabelos, R., Ed.; Academic Press: Oxford, UK, 2019; pp. 191–424.
- Cacabelos, R.; Cacabelos, P.; Carril, J.C. Epigenetics and pharmacoepigenetics of age-related neurodegenerative disorders. In Pharmacoepigenetics, 1st ed.; Cacabelos, R., Ed.; Academic Press: Oxford, UK, 2019; pp. 903–950.
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365.
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069.
- Chu, L.W. Alzheimer’s disease: Early diagnosis and treatment. Hong Kong Med. J. 2012, 18, 228–237.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115.
- Cacabelos, R.; Cacabelos, N.; Carril, J.C. The role of pharmacogenomics in adverse drug reactions. Expert Rev. Clin. Pharmacol. 2019, 12, 407–442.
- Barbe, C.; Jolly, D.; Morrone, I.; Wolak-Thierry, A.; Dramé, M.; Novella, J.L.; Mahmoudi, R. Factors associated with quality of life in patients with Alzheimer’s disease. BMC Geriatr. 2018, 18, 159.
- Ettcheto, M.; Olloquequi, J.; Sánchez-López, E.; Busquets, O.; Cano, A.; Manzine, P.R.; Beas-Zarate, C.; Castro-Torres, R.D.; García, M.L.; Bulló, M.; et al. Benzodiazepines and Related Drugs as a Risk Factor in Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2020, 11, 344.
- Vidoni, E.D.; Kamat, A.; Gahan, W.P.; Ourso, V.; Woodard, K.; Kerwin, D.R.; Binder, E.F.; Burns, J.M.; Cullum, M.; Hydan, L.S.; et al. Baseline Prevalence of Polypharmacy in Older Hypertensive Study Subjects with Elevated Dementia Risk: Findings from the Risk Reduction for Alzheimer’s Disease Study (rrAD). J. Alzheimers Dis. 2020, 77, 175–182.
- Andin, U.; Passant, U.; Gustafson, L.; Englund, E. Alzheimer’s disease (AD) with and without white matter pathology-clinical identification of concurrent cardiovascular disorders. Arch. Gerontol. Geriatr. 2007, 44, 277–286.
- Ibrahim, B.; Suppiah, S.; Piersson, A.D.; Razali, R.M.; Mohamad, M.; Hassan, H.A.; Ibrahim, N. Cardiovascular risk factors of Alzheimer’s disease and other neurocognitive disorders in Malaysia. Med. J. Malays. 2021, 76, 291–297.
- Contois, J.H.; Anamani, D.E.; Tsongalis, G.J. The underlying molecular mechanism of apolipoprotein E polymorphism: Relationships to lipid disorders, cardiovascular disease, and Alzheimer’s disease. Clin. Lab. Med. 1996, 16, 105–123.
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Corzo, L.; Pichel, V.; Kubota, Y. Cerebrovascular risk factors in Alzheimer’s disease: Brain hemodynamics and pharmacogenomic implications. Neurol. Res. 2003, 25, 567–580.
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114.
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746.
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296.
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045.
- Chornenkyy, Y.; Wang, W.X.; Wei, A.; Nelson, P.T. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019, 29, 3–17.
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s disease crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287.
- Cacabelos, R. Pharmacogenomics of Cognitive Dysfunction and Neuropsychiatric Disorders in Dementia. Int. J. Mol. Sci. 2020, 21, 3059.
- Cacabelos, R. Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 673–701.
- Cacabelos, R. Have there been improvement in Alzheimer’s disease drug discovery over the past 5 years? Exp. Opin. Drug Discov. 2018, 13, 523–538.
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed Res. Int. 2020, 2020, 5120230.
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6.
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 2020, 6, e12050.
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Hkan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177.
- Foroutan, N.; Hopkins, R.B.; Tarride, J.E.; Florez, I.D.; Levine, M. Safety and efficacy of active and passive immunotherapy in mild-to-moderate Alzheimer’s disease: A systematic review and network meta-analysis. Clin. Investig. Med. 2019, 42, 53–65.
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J.; et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408, 979–982.
- Herline, K.; Drummond, E.; Wisniewski, T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 2018, 17, 707–721.
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Shrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis. 2019, 6, 63–69.
- Hoskin, J.L.; Sabbagh, M.N.; Al-Hasan, Y.; Decourt, B. Tau immunotherapies for Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 545–554.
- Carrera, I.; Fernandez-Novoa, L.; Aliev, G.; Vigo, C.; Cacabelos, R. Validating Immunotherapy in Alzheimer’s Disease: The EB101 Vaccine. Curr. Pharm. Des. 2016, 22, 849–858.
- Zhang, H.Y.; Zhu, K.; Meng, Y.; Ding, L.; Wang, J.C.; Yin, W.C.; Yan, Y.; Cao, Y.P. Reduction of amyloid beta by Aβ3-10-KLH vaccine also decreases tau pathology in 3×Tg-AD mice. Brain Res. Bull. 2018, 142, 233–240.
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res. Ther. 2018, 10, 115.
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332.
- Lalli, G.; Schott, J.M.; Hardy, J.; De Strooper, B. Aducanumab: A new phase in therapeutic development for Alzheimer’s disease? EMBO Mol. Med. 2021, 13, e14781.
- Mukhopadhyay, S.; Banerjee, D. A Primer on the Evolution of Aducanumab: The First Antibody Approved for Treatment of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 83, 1537–1552.
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701.
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56.
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2016, 2, 169–176.
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95.
- Van de Vrede, L.; Gibbs, D.M.; Koestler, M.; La Joie, R.; Ljubenkov, P.A.; Provost, K.; Soleimani-Meiggoni, D.; Strom, A.; Tsoy, E.; Rabinovici, G.D.; et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement. 2020, 12, e12101.

