Alzheimer’s disease (AD) is a priority health problem with a high cost to society and a large consumption of medical and social resources. The management of AD patients is complex and multidisciplinary. Over 90% of patients suffer from concomitant diseases and require personalized therapeutic regimens to reduce adverse drug reactions (ADRs), drug–drug interactions (DDIs), and unnecessary costs. Men and women show substantial differences in their AD-related phenotypes. Genomic, epigenetic, neuroimaging, and biochemical biomarkers are useful for predictive and differential diagnosis. The most frequent concomitant diseases include hypertension (>25%), obesity (>70%), diabetes mellitus type 2 (>25%), hypercholesterolemia (40%), hypertriglyceridemia (20%), metabolic syndrome (20%), hepatobiliary disorder (15%), endocrine/metabolic disorders (>20%), cardiovascular disorder (40%), cerebrovascular disorder (60–90%), neuropsychiatric disorders (60–90%), and cancer (10%). Over 90% of AD patients require multifactorial treatments with risk of ADRs and DDIs. The implementation of pharmacogenetics in clinical practice can help optimize the limited therapeutic resources available to treat AD and personalize the use of anti-dementia drugs, in combination with other medications, for the treatment of concomitant disorders.
1. Phenotypic Features
In a cohort of randomly-selected patients diagnosed with Alzheimer’s disease (AD) (DSM-V and NINCDS-ADRDA criteria) (n = 2701; age: 67.63 ± 0.19 years; range: 50–96 years) of both sexes (1491 Females (55%); age: 68.26 ± 0.27 years; range: 50–96 years; 1210 Males (45%); age: 66.86 ± 0.28; p < 0.001), retrospectively recruited from the CIBE database (period: 2000–2020), it was investigated sex-related common phenotypes, including biochemistry, hematology, metabolism, hormones, neurotransmitters, cardiovascular and cerebrovascular function, cognition, mood, behavior, and genomic and pharmacogenomic profiles.
In terms of anthropometric parameters, substantial differences are observed between females and males in terms of height, weight, and body-mass index (BMI). Only 28% of patients show normal weight (29% F; 20% M), with obesity in >25% and only 1% with underweight.
Significant differences between females and males are present across many biological parameters, including (i) biochemical (glucose, total-cholesterol, HDL-cholesterol, LDL-cholesterol, triglycerides, urea, creatinine, uric acid, calcium, phosphorous, liver transaminases (ALAT, GGT), alkaline phosphatase, bilirubin, CPK, LDH, ions, iron, ferritin, folate, and vitamin B12), (ii) hormonal (TSH, PRL, ACTH, FSH, LH, estrogen, and testosterone), (iii) hematological (RCB, HCT, Hb, MCV, MCH, WBC, lymphocytes, monocytes, eosinophils, basophils, and platelets), (iv) cognitive, and (v) emotional markers. No differences are found in tumor markers.
Cognitive markers (MMSE, ADAS) indicate than females show worse cognitive performance than males. Cognitive impairment and depression are the first symptoms, which appear in over 90% of LOAD, and 80% and 9%, respectively, in EOAD [1].
Late-life depression is associated with cognitive impairment, and depression is linked with an increased risk for AD. Some overlapping pathogenic substrates (for example, stress, cortisol levels, brain hypoperfusion, neuroinflammation, neurotrophic dysfunction, Aβ accumulation, tauopathic connections, epigenetic factors, and gut microbiota–brain axis) may explain the comorbidity of both clinical entities [2]. Mood disorders are more frequent in women than in men. Over 60% of AD patients show depressive symptoms, which are more severe in women than in men. Likewise, anxiety is also more frequent in females than in males. About 50% of men do not show anxiety, whereas only 30% of women with dementia are free of symptoms in early stages of the disease. Both anxiety and depression fluctuate with the clinical course of the disease [3][4][5][6][7]. Behavioral disorders and psychotic symptoms are also frequent (20–90%) in patients with AD along the clinical course of the disease [3][8][9].
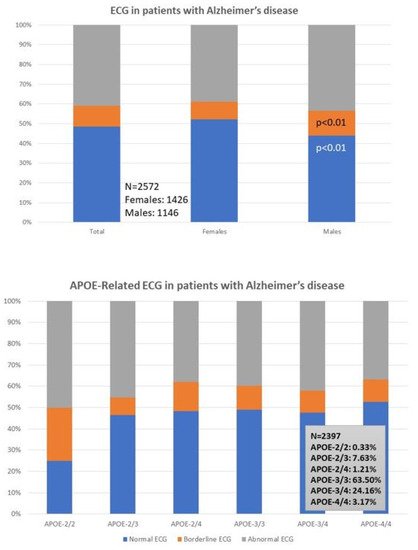
Cardiovascular evaluation (ECG) is abnormal in 40% of the patients (38% F; 43% M). A normal ECG is found more frequently in females (52%) than males (43%; p < 0.01), and a borderline ECG appears more frequently in males (12%) than in females (9%; p < 0.01) (
1. Phenotypic Features
In a cohort of randomly-selected patients diagnosed with AD (DSM-V and NINCDS-ADRDA criteria) (n = 2701; age: 67.63 ± 0.19 years; range: 50–96 years) of both sexes (1491 Females (55%); age: 68.26 ± 0.27 years; range: 50–96 years; 1210 Males (45%); age: 66.86 ± 0.28; p < 0.001), retrospectively recruited from the CIBE database (period: 2000–2020), we investigated sex-related common phenotypes, including biochemistry, hematology, metabolism, hormones, neurotransmitters, cardiovascular and cerebrovascular function, cognition, mood, behavior, and genomic and pharmacogenomic profiles.
In terms of anthropometric parameters, substantial differences are observed between females and males in terms of height, weight, and body-mass index (BMI). Only 28% of patients show normal weight (29% F; 20% M), with obesity in >25% of the cases and only 1% with underweight.
Significant differences between females and males are present across many biological parameters, including (i) biochemical (glucose, total-cholesterol, HDL-cholesterol, LDL-cholesterol, triglycerides, urea, creatinine, uric acid, calcium, phosphorous, liver transaminases (ALAT, GGT), alkaline phosphatase, bilirubin, CPK, LDH, ions, iron, ferritin, folate, and vitamin B12), (ii) hormonal (TSH, PRL, ACTH, FSH, LH, estrogen, and testosterone), (iii) hematological (RCB, HCT, Hb, MCV, MCH, WBC, lymphocytes, monocytes, eosinophils, basophils, and platelets), (iv) cognitive, and (v) emotional markers. No differences are found in tumor markers.
Cognitive markers (MMSE, ADAS) indicate than females show worse cognitive performance than males. Cognitive impairment and depression are the first symptoms, which appear in over 90% of LOAD, and 80% and 9%, respectively, in EOAD cases [25].
Late-life depression is associated with cognitive impairment, and depression is linked with an increased risk for AD. Some overlapping pathogenic substrates (i.e., stress, cortisol levels, brain hypoperfusion, neuroinflammation, neurotrophic dysfunction, Aβ accumulation, tauopathic connections, epigenetic factors, and gut microbiota–brain axis) may explain the comorbidity of both clinical entities [26]. Mood disorders are more frequent in women than in men. Over 60% of AD patients show depressive symptoms, which are more severe in women than in men. Likewise, anxiety is also more frequent in females than in males. About 50% of men do not show anxiety, whereas only 30% of women with dementia are free of symptoms in early stages of the disease. Both anxiety and depression fluctuate with the clinical course of the disease [24,27,28,29,30]. Behavioral disorders and psychotic symptoms are also frequent (20–90%) in patients with AD along the clinical course of the disease [24,31,32].
ECG is abnormal in 40% of the patients (38% F; 43% M). A normal ECG is found more frequently in females (52%) than males (43%; p < 0.01), and a borderline ECG appears more frequently in males (12%) than in females (9%; p < 0.01) (
, upper panel); however, these differences are unrelated to apolipoprotein E (APOE) variants in both sexes (
Figure 1, lower panel). Despite this global perception, and considering the small number of the APOE-2/2 and APOE-4/4 genotypes, APOE-4/4 carriers show abnormal ECGs (52%) more frequently than APOE-3/3 carriers, especially in males (36%) as compared to females (10%); in contrast, homozygous APOE-2/2 carriers exhibit the lowest frequency of abnormal ECG in the whole sample (25%), with twice the number of men (50%) showing abnormal ECG than APOE-2/2 females (25%) (
, lower panel). Despite this global perception, and considering the small number of cases with the APOE-2/2 and APOE-4/4 genotypes, APOE-4/4 carriers show abnormal ECGs (52%) more frequently than APOE-3/3 carriers, especially in males (36%) as compared to females (10%); in contrast, homozygous APOE-2/2 carriers exhibit the lowest frequency of abnormal ECG in the whole sample (25%), with twice the number of men (50%) showing abnormal ECG than APOE-2/2 females (25%) (
lower panel) in patients with Alzheimer’s disease.
2. Biomarkers
In addition to conventional clinical markers, which was allowed to make a differential diagnosis and assess the possibility of concomitant diseases, the most useful biomarkers for a predictive diagnosis or diagnostic confirmation of antemortem AD are genomic markers, epigenetic biomarkers, neurotransmitters, and levels of Aβ/tau in the brain (PET Scan) and/or in body fluids [10][11][12][13][14][15][16][17][18][19].
2.1. Genomic Markers
Over 600 human genes are associated with AD [20][21][22]. Mutations in the amyloid precursor protein (APP) (>50 different mutations), presenilin 1 (PSEN1 > 300 mutations), and presenilin-2 (PSEN2 > 40 mutations) genes are present in a number of AD (5–10%), and induce brain amyloidopathy. Microtubule-associated protein tau (MAPT) gene mutations (>100), also present in some patients with AD, may cause brain tauopathies (for example, frontotemporal dementia, Pick’s disease) [23][24][25]. Both conditions (amyloidopathy and tauopathy) are the two dominant pathogenic hypotheses in AD [26][27].
APP mutations can cause EOAD, with increased Aβ levels or Aβ fibrillogenesis, while some coding variants (APP A673T) may be protective with reduced Aβ levels [25]. EOAD dominant mutations tend to occur in the APP coding region or in the presenilin-related catalytic site of γ-secretase whose protease dysfunction is responsible for the abnormal process of APP cleavage and consequent accumulation of Aβ in senile plaques and vessels. In >40% AD, the presence of the apolipoprotein E4 (APOE-4) allele is the most important risk factor, involved in the impairment of Aβ clearance from brain tissue, in atherosclerosis and in hypoperfusion. Most immunotherapeutic interventions with Aβ antibodies (aducanumab, solanezumab, and crenezumab) attempt to halt the amyloidogenic process and slow-down cognitive deterioration in mild-AD or in presymptomatic ones with demonstrated genomic dysfunction [26]. In addition to these primary pathogenic genes, many other genes have been associated with AD in next-generation sequencing (NGS) and genome-wide association studies (GWAS) in different populations [21][22][28][29][30][31][32].
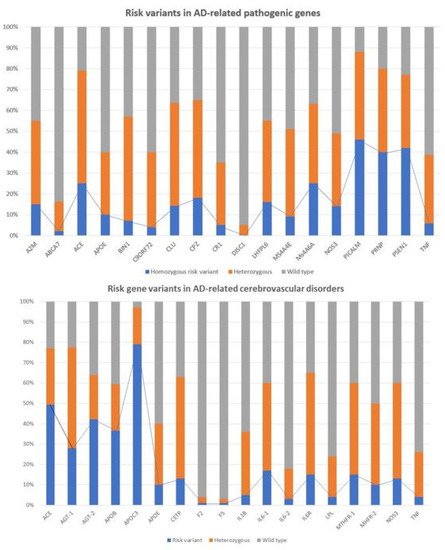
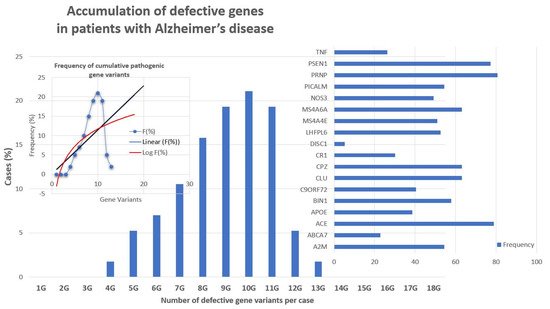
Recently, it was showed that multiple genetic defects can accumulate in the same of AD, conditioning its phenotypic characteristics. In a genomic panel of 18 AD-related pathogenic genes and genotypes related to cerebrovascular disorders () in patients with Alzheimer’s disease.
2. Biomarkers
In addition to conventional clinical markers, which allow us to make a differential diagnosis and assess the possibility of concomitant diseases, the most useful biomarkers for a predictive diagnosis or diagnostic confirmation of antemortem AD are genomic markers, epigenetic biomarkers, neurotransmitters, and levels of Aβ/tau in the brain (PET Scan) and/or in body fluids [37,38,39,40,41,42,43,44,45,46].
2.1. Genomic Markers
Over 600 human genes are associated with AD [5,47,48]. Mutations in the amyloid precursor protein (APP) (>50 different mutations), presenilin 1 (PSEN1 > 300 mutations), and presenilin-2 (PSEN2 > 40 mutations) genes are present in a number of AD cases (5–10%), and induce brain amyloidopathy. Microtubule-associated protein tau (MAPT) gene mutations (>100), also present in some patients with AD, may cause brain tauopathies (e.g., frontotemporal dementia, Pick’s disease) [49,50,51]. Both conditions (amyloidopathy and tauopathy) are the two dominant pathogenic hypotheses in AD [52,53].
APP mutations can cause EOAD, with increased Aβ levels or Aβ fibrillogenesis, while some coding variants (APP A673T) may be protective with reduced Aβ levels [51]. EOAD dominant mutations tend to occur in the APP coding region or in the presenilin-related catalytic site of γ-secretase whose protease dysfunction is responsible for the abnormal process of APP cleavage and consequent accumulation of Aβ in senile plaques and vessels. In >40% AD cases, the presence of the apolipoprotein E4 (APOE-4) allele is the most important risk factor, involved in the impairment of Aβ clearance from brain tissue, in atherosclerosis and in hypoperfusion. Most immunotherapeutic interventions with Aβ antibodies (aducanumab, solanezumab, and crenezumab) attempt to halt the amyloidogenic process and slow-down cognitive deterioration in mild-AD or in presymptomatic cases with demonstrated genomic dysfunction [52]. In addition to these primary pathogenic genes, many other genes have been associated with AD in next-generation sequencing (NGS) and genome-wide association studies (GWAS) in different populations [47,48,54,55,56,57,58].
Recent studies show that multiple genetic defects can accumulate in the same case of AD, conditioning its phenotypic characteristics. In a genomic panel of 18 AD-related pathogenic genes (Table 2) and genotypes related to cerebrovascular disorders (
), it can be verified that >60% of patients are carriers of more than 10 pathogenic variants. Pathogenic gene variants (
) and cerebrovascular risk gene variants (
lower panel) associated with Alzheimer’s disease.
) associated with Alzheimer’s disease. See Table 2 and Table 3 and Abbreviations for gene identification and SNPs of risk.
The genes that most frequently accumulate pathogenic variants (>50%) in cases of AD are the following: PRNP (80.70%), ACE (78.94%), PSEN1 (77.19%), CLU (63.15%), CPZ (63.15%), MS4A6A (63.15%), BIN1 (57.89%), A2M (54.38%), PICALM (54.38%), LHFPL6 (52.63%), and MS4A4E (50.87%) (
Figure 3. Accumulation of defective pathogenic gene variants in patients with Alzheimer’s disease.
2.2. Epigenetic Markers
Various epigenetic aberrations are associated with AD pathogenesis, including hypomethylation/hypermethylation in the promoters of pathogenic genes, alterations in histones, and changes in the linear and three-dimensional structure of nuclear chromatin, as well as profound alterations in microRNAs (miRNAs) that regulate gene expression in the cellular cytoplasm. Some of these epigenetic alterations have been proposed as potential biomarkers of AD [50][51][52][53][54][55][56][57].
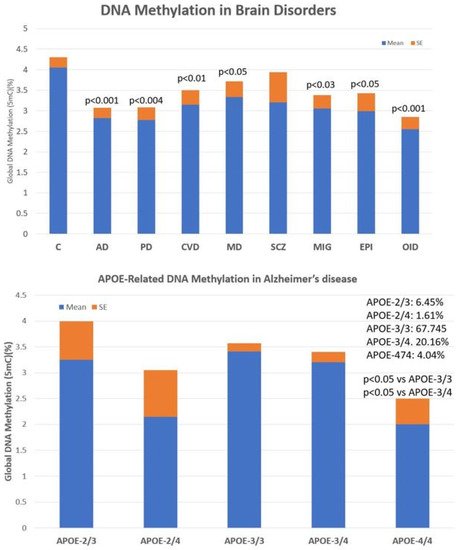
The main problems observed with the use of epigenetic biomarkers in AD are their variability and lack of specificity. Changes in global DNA methylation are very sensitive and appear diminished in a multitude of central nervous system (CNS) diseases, such as AD (p < 0.001), Parkinson’s disease (p < 0.004), cerebrovascular disorders and stroke (p < 0.01), major depression (p < 0.05), migraine (p < 0.03), epilepsy (p < 0.05), and intellectual organic disability (OID) (p < 0.001), and to a lesser extent in schizophrenia ( Accumulation of defective pathogenic gene variants in patients with Alzheimer’s disease.
The pathogenic load of APOE-4 affects 35–40% of cases, with significant phenotypic consequences (Table 2, Table 3 and Table 4). APOE-4/4 carriers tend to show an earlier age-at-onset in >80% of the cases; lower peripheral ApoE, nitric oxide, histamine, Aβ, HDL-cholesterol, and triglyceride levels; higher levels of total cholesterol and LDL-cholesterol; more pronounced brain atrophy and slower brain bioelectrical activity; more severe brain hemodynamic dysfunction represented by hypoperfusion, reduced brain blood flow velocity and increased pulsatility and resistance indices; increased lymphocyte apoptosis; faster cognitive deterioration; more frequent metabolic disorders, cardiovascular disorders, hypertension, atherosclerosis, liver metabolism dysfunction, behavioral disturbances, and alterations in circadian rhythm patterns; and a poor response to conventional treatments [5,11,12,13,22,48,59,60,61,62,63,64,65,66,67,68,69,70,71].
2.2. Epigenetic Markers
Various epigenetic aberrations are associated with AD pathogenesis, including hypomethylation/hypermethylation in the promoters of pathogenic genes, alterations in histones, and changes in the linear and three-dimensional structure of nuclear chromatin, as well as profound alterations in microRNAs (miRNAs) that regulate gene expression in the cellular cytoplasm. Some of these epigenetic alterations have been proposed as potential biomarkers of AD [72,73,74,75,76,77,78,79].
The main problems observed with the use of epigenetic biomarkers in AD are their variability and lack of specificity. Changes in global DNA methylation are very sensitive and appear diminished in a multitude of central nervous system (CNS) diseases, such as AD (p < 0.001), Parkinson’s disease (p < 0.004), cerebrovascular disorders and stroke (p < 0.01), major depression (p < 0.05), migraine (p < 0.03), epilepsy (p < 0.05), and intellectual organic disability (OID) (p < 0.001), and to a lesser extent in schizophrenia (
, upper panel). These values are very sensitive to therapeutic interventions but unreliable as predictive or diagnostic values. The low diagnostic value of DNA methylation is compensated for by the exquisite sensitivity of this biomarker that responds in a highly sensitive manner to the therapeutic response of each patient. In addition, what appears to be important in AD is that global DNA methylation shows an APOE-dependent pattern. APOE-4 carriers tend to show a more severe DNA hypomethylation pattern than patients carrying the APOE-3 allele, which is aggravated in parallel with the degree of cognitive impairment (
Global DNA methylation in patients with central nervous system disorders (
) and APOE-related DNA methylation in patients with Alzheimer’s disease (
lower panel). C: Control; AD: Alzheimer’s disease; PD: Parkinson’s disease; CVD: Cerebrovascular disorder; MD: Major Depression; SCZ: Schizophrenia and psychotic syndromes; MIG: Migraine; EPI: Epilepsy; OID: Organic Intellectual Disability.
Epigenetic biomarkers can also help in the personalization of anti-AD treatments (Pharmacoepigenetics) [58][59][60][61] and serve as a guide in the search for epigenetic drugs with prophylactic and/or therapeutic action in the treatment of AD [50][59][60][61].
2.3. Neurotransmitters
Conventionally, the main neurotransmitter affected in AD is acetylcholine. However, premature neuronal death alters the levels of many other essential neurotransmitters for the normal functioning of the CNS [62][63][64].
Deficits in noradrenaline, dopamine, serotonin, histamine, GABA, glutamate, and various neuropeptides (GRF, CRF, somatostatin, and vasopressin) are particularly relevant, the alteration of which can lead to AD-related neuropsychiatric disorders. However, none of these biomarkers are sufficiently sensitive or specific to AD ( ). C: Control; AD: Alzheimer’s disease; PD: Parkinson’s disease; CVD: Cerebrovascular disorder; MD: Major Depression; SCZ: Schizophrenia and psychotic syndromes; MIG: Migraine; EPI: Epilepsy; OID: Organic Intellectual Disability.
Epigenetic biomarkers can also help in the personalization of anti-AD treatments (Pharmacoepigenetics) [80,81,82,83] and serve as a guide in the search for epigenetic drugs with prophylactic and/or therapeutic action in the treatment of AD [72,81,82,83].
2.3. Neurotransmitters
Conventionally, the main neurotransmitter affected in AD is acetylcholine. However, premature neuronal death alters the levels of many other essential neurotransmitters for the normal functioning of the CNS [84,85,86].
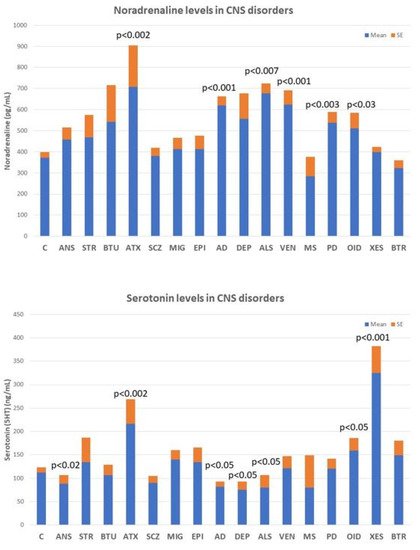
Deficits in noradrenaline, dopamine, serotonin, histamine, GABA, glutamate, and various neuropeptides (GRF, CRF, somatostatin, and vasopressin) are particularly relevant, the alteration of which can lead to AD-related neuropsychiatric disorders. However, none of these biomarkers are sufficiently sensitive or specific to AD (
), although their quantification in CSF or blood is useful for monitoring brain damage and/or the efficacy or ineffectiveness of the treatments the patient receives. ) in central nervous system disorders. C: Control; ANS: Anxiety; STR: Stroke; BTU: Brain tumors; ATX: Ataxia; SCZ; Schizophrenia and psychosis; MIG: Migraine; EPI: Epilepsy; AD: Alzheimer’s disease; DEP: Depression; ALS: Amyotrophic Lateral Sclerosis; VEN: Vascular encephalopathy; MS: Multiple Sclerosis; PD: Parkinson’s disease; OID: Organic Intellectual Disability; XES: Xenoestrogenic syndrome; and BTR: Brain trauma.
Noradrenaline levels in the blood increase significantly in most neurodegenerative diseases, including ataxic syndromes (p < 0.002), AD (p < 0.001), amyotrophic lateral sclerosis (ALS) (p < 0.007), and Parkinson’s disease (p < 0.003), as well as in vascular encephalopathies (p < 0.001) and in OID (
, upper panel). In contrast, serotonin levels tend to decrease in anxiety (p < 0.02), AD (p < 0.05), depression (p < 0.05), and ALS (p < 0.05), and show high levels in ataxic syndromes (p < 0.002), OID (p < 0.05), and in xenoestrogenic syndrome (p < 0.001), a novel clinical entity present in women with chronic use of contraceptives or hormone replacement therapy (HRT) (
Figure 5, lower panel).
2.4. Aβ/Tau Levels
The most popular biomarkers for AD in body fluids (CSF, plasma) are the quantification of amyloid-β (Aβ42), total tau (T-tau), and phosphorylated tau (P-tau) in the CSF (AD CSF profile: decreased Aβ42 levels together with increased T-tau and P-tau levels). Other CSF markers (synaptotagmin, rab3a, SNAP-25, and neurogranin) are also altered [13][14]. However, the heterogeneity of AD and the inconvenience of having to perform a lumbar puncture to obtain CSF do not allow these biomarkers to reach high quotas of sensitivity and specificity, nor generalized use.
3. Concomitant Disorders and Phenotype-Modifying Treatments
Most patients with dementia (>90%) require multifactorial treatments for the management of concomitant disorders and/or neuropsychiatric symptoms associated with dementia. The chronic administration of drugs from different categories increases the risk of ADRs and DDIs [65][66][67][68].
The most frequent concomitant disorders in AD are the following: systolic hypertension (21%), diastolic hypertension (28%), obesity (>70%), diabetes mellitus type 2 (26%), hypercholesterolemia (40%), hypertriglyceridemia (20%), hyperuricemia (6%), metabolic syndrome (20%), transaminitis (11%), hyperbilirubinemia (15%), endocrine disorders (5%), iron deficiency anemia (7%), folate deficit (17%), vitamin B12 deficit (10%), cardiovascular disorder (40%), cerebrovascular disorder (>90% in patients over 80 years of age), anxiety (60%), depression (65%), behavioral disorders (20–90%), and cancer (10%).
Cardiovascular risk factors (hypertension, hypercholesterolemia, and dyslipidemia) and ECG abnormalities are more frequent in males than in females. Hypertension is present in 21%. Systolic blood pressure is similar in females and males, but diastolic blood pressure is significantly higher in males than in females (p < 0.001). Cholesterol levels (Total, LDL) are higher in men and HDL-cholesterol and triglyceride levels are more elevated in females. Brain damage and increased cognitive deterioration are currently associated with cardiovascular disorders and blood pressure changes in AD [69][70]. APOE-4 carriers with dementia also exhibit cardiovascular disorders, atherosclerosis, and cerebrovascular damage [16][17][18][20][34][38][39][40][41][42][43][44][71]. Lipid metabolism disorders contribute to the cerebrovascular component of AD. Abnormalities in cholesterol metabolism and dysfunction of lipid rafts in cell membranes and arteriosclerosis are pathogenically relevant for cerebral ischemia and hypoperfusion, and accelerate premature neuronal death in patients who are predisposed to AD [17][72][73][74][75]. In contrast, the epidemiological link between diabetes and AD appears to be circumstantial, with no apparent pathogenic implications beyond the deleterious effects of hyperglycemia on brain function [76][77][78].
As a consequence of all these concomitant pathologies, patients with dementia consume a wide variety of drugs whose side-effects contribute to accelerating the degenerative process and cognitive decline. Of special importance are cardiovascular agents, statins, antidiabetics, antihypertensive drugs, analgesics, diuretics, bronchodilators, antirheumatics, and various categories of psychotropic drugs (neuroleptics, antidepressants, anxiolytics, hypnotics, and sedatives). The correct administration of these drugs requires a personalized therapeutic intervention, together with conventional anti-dementia treatments [65][79][80].
Combination treatments applied under pharmacogenetic guidance indicate that alterations in biochemical, hematological, and metabolic parameters affect drug efficacy and safety. Concerning cognitive function and neuropsychiatric disorders treated with multifactorial regimes, females and males respond differentially to treatment, showing a moderate improvement in cognition during the first year of treatment and significant improvements in anxiety and depression.
Several pharmacogenetic ones agree that APOE-4 carriers respond poorly to conventional treatments, while APOE-3 carriers tend to respond better to different therapeutic regimens. Similarly, normal metabolizers (NMs) and intermediate metabolizers (IMs) associated with the different genotypes of the CYP2D6, CYP2C9, and CYP2C19 genes show a better therapeutic response than poor metabolizers (PMs) and ultra-rapid metabolizers (UMs) to treatments with anti-dementia drugs and psychotropic drugs to regulate aberrant behaviors [3][38][39][40][41][42][43][44][45].
4. Alzheimer’s Disease Therapeutics and Drug Development
For the past 50 years, the major focus of pharmacological development in AD has been cognitive enhancers. The introduction of acetylcholinesterase inhibitors (AChEIs) in the early 1990s was the first option to restore cholinergic neurotransmission after the identification of a selective cholinergic deficit in the basal forebrain and neuronal loss in neocortex and hippocampus. Tacrine (9-amino-1,2,3,4-tetrahydroacridine) was the first AChEI introduced in 1993 for the treatment of AD. The Chinese product Huperzine A was approved in 1994.
A new generation of AChEIs (donepezil, galantamine, and rivastigmine) was introduced years later. In 2003, the FDA approved memantine, an N-methyl-D-aspartate (NMDA) glutamate receptor partial inhibitor. Since then, no new FDA-approved drugs for AD were reported until the recent approval of the antibody aducanumab in 2021 [80][81].
The main categories of drugs known during the last two decades as candidates for the treatment of AD were heterogeneous and include the following: neurotransmitter enhancers (11.38%), anti-amyloid agents (13.30%), anti-tau agents (2.03%), multi-target drugs (8.11%), novel synthetic drugs (8.13%), neuroprotective peptides (1.25%), old repository drugs (11.77%), anti-inflammatory drugs (1.20%), and a large number of natural products and derivatives (25.58%). Novel categories of therapeutic intervention (stem cell therapy, nanocarriers/nanotherapeutics) and combination treatments have also been challenged [81]. Over 2000 new AChEIs, some of them with dual inhibitory activity on acetylcholinesterase and butyrylcholinesterase, have been identified, as well as over 150 multi-target drugs [81][82]. Approximately 15% of pharmacological ones searching for anti-AD drugs focused on anti-amyloidogenic strategies (immunotherapy, APP modulators, α-secretase modulators, β-secretase (BACE) inhibitors, ϒ-secretase modulators, Aβ aggregation inhibitors, Notch inhibitors, β-sheet breakers, and Aβ scavengers), with notorious failures [81]. For the past two years, an increase in the number of disease-modifying agents targeting non-amyloid or tau pathogenic cascades has been observed [81][82][83][84].
4.1. Immunotherapy
Since the early 2000s, several categories of vaccines and immunotherapeutic procedures have been developed for the treatment of AD, following the pioneering ones of Schenk and coworkers in 1999 [85]. For two decades, millions of dollars have been invested in passive and active immunotherapy in experimental AD models and in clinical trials. About 1000 papers on AD immunotherapy were published (85% anti-Aβ and 15% anti-tau) prior to the FDA approval of aducanumab as an immunotherapeutic strategy in mild-AD [83][86].
Active immunization ones demonstrate that presymptomatic immunization of PDAPP transgenic mice, overexpressing human mutant APP (Phe717Val), prevents Aβ-plaque formation, astrogliosis, and neuritic dystrophy. Immunization with Aβ42 reduces the extent and progression of AD-related neurodegeneration and improves cognition in the TgCRND8 murine model of AD [85][86][87][88]. Over 400 experimental ones with different immunization procedures were able to replicate these results. However, clinical ones did not show the same efficacy and initial clinical trials with the Aβ vaccine AN1792 had to be discontinued. This was because of severe undesirable effects, such as acute meningoencephalitis or microhemorrhagic lesions in the brain of some immunized patients. Some of these ADRs were attributed to a T-cell-mediated pro-inflammatory response; other mechanistic causes of brain damage in patients treated with active vaccines remain elusive. Improvements in immunization processes and vaccine preparations in recent years have made it possible to obviate many of these drawbacks and avoid adverse effects with the second-generation Aβ-active immunotherapies, anti-tau immunotherapies, and anti-Aβ monoclonal antibodies targeting Aβ epitopes [89][90][91][92][93][94]. Other modalities of immunization, such as dual vaccines (EB101) [91], Aβ3-10-KLH vaccine [92], or active full-length DNA-Aβ42 trimer immunization [93], also reduce amyloidogenic and tauopathic markers in transgenic animals [94].
On 7 June 2021, aducanumab was formally approved by the FDA as a putative disease-modifying antibody for the treatment of AD [95][96][97][98][99][100]. Aducanumab penetrates into the brain, binds parenchymal Aβ, selectively targets aggregated Aβ, and dose-dependently reduces Aβ levels, with parallel slowing of cognitive decline [96][97][98][99]. Vasogenic edema is one of the major ADRs of aducanumab and other injectable antibodies (BAN2401), especially in APOE-4 carriers [100][101].
, lower panel).
2.4. Aβ/Tau Levels
The most popular biomarkers for AD in body fluids (CSF, plasma) are the quantification of amyloid-β (Aβ42), total tau (T-tau), and phosphorylated tau (P-tau) in the CSF (AD CSF profile: decreased Aβ42 levels together with increased T-tau and P-tau levels). Other CSF markers (synaptotagmin, rab3a, SNAP-25, and neurogranin) are also altered [40,41]. However, the heterogeneity of AD and the inconvenience of having to perform a lumbar puncture to obtain CSF do not allow these biomarkers to reach high quotas of sensitivity and specificity, nor generalized use.
3. Concomitant Disorders and Phenotype-Modifying Treatments
Most patients with dementia (>90%) require multifactorial treatments for the management of concomitant disorders and/or neuropsychiatric symptoms associated with dementia. The chronic administration of drugs from different categories increases the risk of ADRs and DDIs [17,87,88,89].
The most frequent concomitant disorders in AD cases are the following: systolic hypertension (21%), diastolic hypertension (28%), obesity (>70%), diabetes mellitus type 2 (26%), hypercholesterolemia (40%), hypertriglyceridemia (20%), hyperuricemia (6%), metabolic syndrome (20%), transaminitis (11%), hyperbilirubinemia (15%), endocrine disorders (5%), iron deficiency anemia (7%), folate deficit (17%), vitamin B12 deficit (10%), cardiovascular disorder (40%), cerebrovascular disorder (>90% in patients over 80 years of age), anxiety (60%), depression (65%), behavioral disorders (20–90%), and cancer (10%).
Cardiovascular risk factors (hypertension, hypercholesterolemia, and dyslipidemia) and ECG abnormalities are more frequent in males than in females. Hypertension is present in 21% of the cases. Systolic blood pressure is similar in females and males, but diastolic blood pressure is significantly higher in males than in females (p < 0.001). Cholesterol levels (Total, LDL) are higher in men and HDL-cholesterol and triglyceride levels are more elevated in females. Brain damage and increased cognitive deterioration are currently associated with cardiovascular disorders and blood pressure changes in AD [90,91]. APOE-4 carriers with dementia also exhibit cardiovascular disorders, atherosclerosis, and cerebrovascular damage [5,12,43,44,45,60,61,62,63,64,65,66,92]. Lipid metabolism disorders contribute to the cerebrovascular component of AD. Abnormalities in cholesterol metabolism and dysfunction of lipid rafts in cell membranes and arteriosclerosis are pathogenically relevant for cerebral ischemia and hypoperfusion, and accelerate premature neuronal death in patients who are predisposed to AD [44,93,94,95,96]. In contrast, the epidemiological link between diabetes and AD appears to be circumstantial, with no apparent pathogenic implications beyond the deleterious effects of hyperglycemia on brain function [97,98,99].
As a consequence of all these concomitant pathologies, patients with dementia consume a wide variety of drugs whose side-effects contribute to accelerating the degenerative process and cognitive decline. Of special importance are cardiovascular agents, statins, antidiabetics, antihypertensive drugs, analgesics, diuretics, bronchodilators, antirheumatics, and various categories of psychotropic drugs (neuroleptics, antidepressants, anxiolytics, hypnotics, and sedatives). The correct administration of these drugs requires a personalized therapeutic intervention, together with conventional anti-dementia treatments [15,17,100].
Combination treatments applied under pharmacogenetic guidance indicate that alterations in biochemical, hematological, and metabolic parameters affect drug efficacy and safety. Concerning cognitive function and neuropsychiatric disorders treated with multifactorial regimes, females and males respond differentially to treatment, showing a moderate improvement in cognition during the first year of treatment and significant improvements in anxiety and depression.
Several pharmacogenetic studies agree that APOE-4 carriers respond poorly to conventional treatments, while APOE-3 carriers tend to respond better to different therapeutic regimens. Similarly, normal metabolizers (NMs) and intermediate metabolizers (IMs) associated with the different genotypes of the CYP2D6, CYP2C9, and CYP2C19 genes show a better therapeutic response than poor metabolizers (PMs) and ultra-rapid metabolizers (UMs) to treatments with anti-dementia drugs and psychotropic drugs to regulate aberrant behaviors [24,60,61,62,63,64,65,66,67].
4. Alzheimer’s Disease Therapeutics and Drug Development
For the past 50 years, the major focus of pharmacological development in AD has been cognitive enhancers. The introduction of acetylcholinesterase inhibitors (AChEIs) in the early 1990s was the first option to restore cholinergic neurotransmission after the identification of a selective cholinergic deficit in the basal forebrain and neuronal loss in neocortex and hippocampus. Tacrine (9-amino-1,2,3,4-tetrahydroacridine) was the first AChEI introduced in 1993 for the treatment of AD. The Chinese product Huperzine A was approved in 1994.
A new generation of AChEIs (donepezil, galantamine, and rivastigmine) was introduced years later. In 2003, the FDA approved memantine, an N-methyl-D-aspartate (NMDA) glutamate receptor partial inhibitor. Since then, no new FDA-approved drugs for AD were reported until the recent approval of the antibody aducanumab in 2021 [100,101].
The main categories of drugs studied during the last two decades as candidates for the treatment of AD were heterogeneous and include the following: neurotransmitter enhancers (11.38%), anti-amyloid agents (13.30%), anti-tau agents (2.03%), multi-target drugs (8.11%), novel synthetic drugs (8.13%), neuroprotective peptides (1.25%), old repository drugs (11.77%), anti-inflammatory drugs (1.20%), and a large number of natural products and derivatives (25.58%). Novel categories of therapeutic intervention (stem cell therapy, nanocarriers/nanotherapeutics) and combination treatments have also been challenged [101]. Over 2000 new AChEIs, some of them with dual inhibitory activity on acetylcholinesterase and butyrylcholinesterase, have been identified, as well as over 150 multi-target drugs [101,102]. Approximately 15% of pharmacological studies searching for anti-AD drugs focused on anti-amyloidogenic strategies (immunotherapy, APP modulators, α-secretase modulators, β-secretase (BACE) inhibitors, ϒ-secretase modulators, Aβ aggregation inhibitors, Notch inhibitors, β-sheet breakers, and Aβ scavengers), with notorious failures [101]. For the past two years, an increase in the number of disease-modifying agents targeting non-amyloid or tau pathogenic cascades has been observed [101,102,103,104].
Immunotherapy
Since the early 2000s, several categories of vaccines and immunotherapeutic procedures have been developed for the treatment of AD, following the pioneering studies of Schenk and coworkers in 1999 [105]. For two decades, millions of dollars have been invested in passive and active immunotherapy in experimental AD models and in clinical trials. About 1000 papers on AD immunotherapy were published (85% anti-Aβ and 15% anti-tau) prior to the FDA approval of aducanumab as an immunotherapeutic strategy in mild-AD [103,106].
Active immunization studies demonstrate that presymptomatic immunization of PDAPP transgenic mice, overexpressing human mutant APP (Phe717Val), prevents Aβ-plaque formation, astrogliosis, and neuritic dystrophy. Immunization with Aβ42 reduces the extent and progression of AD-related neurodegeneration and improves cognition in the TgCRND8 murine model of AD [105,106,107,108]. Over 400 experimental studies with different immunization procedures were able to replicate these results. However, clinical studies did not show the same efficacy and initial clinical trials with the Aβ vaccine AN1792 had to be discontinued. This was because of severe undesirable effects, such as acute meningoencephalitis or microhemorrhagic lesions in the brain of some immunized patients. Some of these ADRs were attributed to a T-cell-mediated pro-inflammatory response; other mechanistic causes of brain damage in patients treated with active vaccines remain elusive. Improvements in immunization processes and vaccine preparations in recent years have made it possible to obviate many of these drawbacks and avoid adverse effects with the second-generation Aβ-active immunotherapies, anti-tau immunotherapies, and anti-Aβ monoclonal antibodies targeting Aβ epitopes [109,110,111,112,113,114]. Other modalities of immunization, such as dual vaccines (EB101) [111], Aβ3-10-KLH vaccine [112], or active full-length DNA-Aβ42 trimer immunization [113], also reduce amyloidogenic and tauopathic markers in transgenic animals [114].
On 7 June 2021, aducanumab was formally approved by the FDA as a putative disease-modifying antibody for the treatment of AD [115,116,117,118,119,120]. Aducanumab penetrates into the brain, binds parenchymal Aβ, selectively targets aggregated Aβ, and dose-dependently reduces Aβ levels, with parallel slowing of cognitive decline [116,117,118,119]. Vasogenic edema is one of the major ADRs of aducanumab and other injectable antibodies (BAN2401), especially in APOE-4 carriers [120,121].