Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carsten Carlberg | -- | 2316 | 2022-04-12 09:47:08 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Carlberg, C. Vitamin D Signaling. Encyclopedia. Available online: https://encyclopedia.pub/entry/21622 (accessed on 27 June 2026).
Carlberg C. Vitamin D Signaling. Encyclopedia. Available at: https://encyclopedia.pub/entry/21622. Accessed June 27, 2026.
Carlberg, Carsten. "Vitamin D Signaling" Encyclopedia, https://encyclopedia.pub/entry/21622 (accessed June 27, 2026).
Carlberg, C. (2022, April 12). Vitamin D Signaling. In Encyclopedia. https://encyclopedia.pub/entry/21622
Carlberg, Carsten. "Vitamin D Signaling." Encyclopedia. Web. 12 April, 2022.
Copy Citation
The vitamin D metabolite 1α,25-dihydroxyvitamin D3 is the natural, high-affinity ligand of the transcription factor vitamin D receptor (VDR). In many tissues and cell types, VDR binds in a ligand-dependent fashion to thousands of genomic loci and modulates, via local chromatin changes, the expression of hundreds of primary target genes. Thus, the epigenome and transcriptome of VDR-expressing cells is directly affected by vitamin D. Vitamin D target genes encode for proteins with a large variety of physiological functions, ranging from the control of calcium homeostasis, innate and adaptive immunity, to cellular differentiation.
vitamin D
VDR
target genes
1. VDR: A Transcription Factor Activated by Vitamin D
Transcription factors are proteins that are able to bind sequence-specifically to genomic DNA and interact with other nuclear proteins, which modulates the activity of RNA polymerase II and mRNA production [1][2]. Some of the approximately 1600 transcription factors encoded by the human genome are constitutively active and regulated primarily by their expression, while most of them are activated by extra- and intracellular signals. A few of these signal-dependent transcription factors are located either in a latent form in the cytosol and activated through translocation into the nucleus, or—most of them—are found in the nucleus and modulated in their activity by post-translational modifications, such as phosphorylation or acetylation. Furthermore, some members of the nuclear receptor superfamily have an additional mechanism of activation, which is a ligand-induced conformational change on the surface of their ligand-binding domain (LBD) [3][4].
The inner surface of VDR’s LBD forms a ligand-binding pocket, where 40, mostly non-polar, amino acids snugly enclose the molecule 1,25(OH)2D3, so that it binds with an affinity of 0.1 nM [5]. This is a very high affinity, even in comparison with other nuclear receptors [6]. Ligand binding changes VDR’s interaction profile with many of the more than 50 nuclear proteins that have been reported to cooperate with the receptor [7]. Some of these VDR-interacting proteins function as co-repressors, such as NCOR1 (nuclear receptor corepressor 1) [8] or COPS2 (COP9 signalosome subunit 2, also called ALIEN) [9], co-activators of the NCOA (nuclear receptor coactivator) family [10], or members of the Mediator complex, such as MED1 [11][12][13] (Figure 1). Other VDR partner proteins are chromatin-modifying enzymes, such as histone acetyltransferases (HATs) [10], histone deacetylases (HDACs) [9], lysine demethylases, such as KDM6B [14] and KDM1A [15] or chromatin remodeling proteins, such as BRD (bromodomain-containing) 7 and 9 [16]. The large variety of its protein interaction partners suggests that VDR is a dynamic member of a large nuclear protein complex [17].
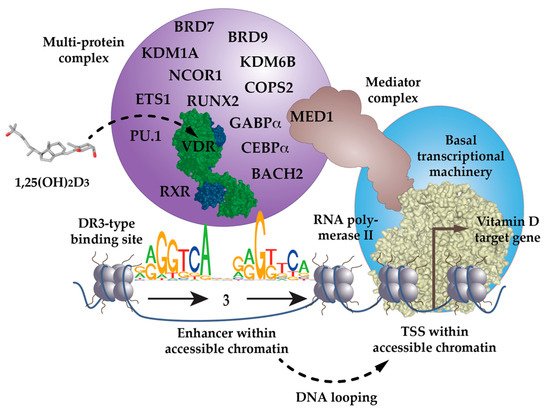
Figure 1. VDR as the key ligand-inducible component of a multi-protein complex. VDR is a part of a multi-protein complex that, e.g., contains co-receptors (RXR), pioneer factors (PU.1, CEBPα, GABPα, ETS1, RUNX2, BACH2), chromatin modifiers (KDM1A, KDM6B), chromatin remodelers (BRD7, BRD9), co-activators (MED1) and co-repressors (NCOR1, COPS2). The complex is activated through the binding of 1,25(OH)2D3 to VDR and attaches preferentially to DR3-type binding sites within enhancer regions. The mediator complex connects the activated VDR complex with the RNA polymerase II waiting on transcription start site (TSS) regions of vitamin D target genes. In most cases the linear distance of enhancer and TSS region are multiple kb, so that the intervening genomic DNA forms a regulatory loop. In this way the expression of the vitamin D target genes is either increased or decreased.
The low percentage of DR3-binding VDR complexes detected by ChIP-seq also suggests that there are a number of scenarios in which VDR acts independently of RXR. VDR may use other nuclear proteins as alternative cooperative binding partners on genomic DNA [18][19][20] or may bind indirectly to DNA, such as “backpack”, to other transcription factors [21]. For example, below VDR ChIP-seq peak binding sites for the pioneer transcription factor PU.1 (purine-rich box-1) are enriched [22]. In fact, in THP-1 monocytic leukemia cells, the presence of PU.1 is observed on two-thirds of VDR’s genomic binding sites [23]. This makes sense, since PU.1, VDR and the pioneer factor CEBPα (CCAAT enhancer binding protein alpha) are the key transcription factors directing the differentiation of myeloid progenitor cells into monocytes and granulocytes, during the process of hematopoiesis [24]. Interestingly, THP-1 cells CEBPα [25], GABPα (GA-binding protein transcription factor alpha) [26] and ETS1 (ETS proto-oncogene 1, transcription factor) [27] also co-locate with VDR binding sites and act as pioneer factors for vitamin D signaling (Figure 1). Furthermore, in osteoblasts, vitamin D signaling is enhanced by the pioneer factors CEBPα and RUNX2 (RUNX family transcription factor 2) [28], while in T cells, this is mediated by the transcription factor BACH2 (BTB domain and CNC homolog 2) [29]. Thus, VDR uses help from RXR, but also from many other transcription factors, in order to form functional complexes with genomic DNA.
2. Vitamin D Target Gene Regulation in the Context of Chromatin
TADs are far larger loops of genomic DNA than regulatory loops, with a size of hundreds of kb to a few Mb [30]. They subdivide the human genome into at least 2000 units, which are functionally insulated from each other [31] (Figure 2). The borders of TADs are defined by the binding of the chromatin-organizing protein CTCF (CCCTC-binding factor) [32][33], forming together with cohesin and other proteins, so-called TAD anchors [34]. The interaction of the VDR-bound enhancers with genomic regions outside of a TAD are prevented by these insulating TAD borders. This is the reason why genes are regulated almost exclusively by enhancers that are located within the same TAD. This also implies that the linear distance between VDR-bound enhancers and TSS region(s) cannot be larger than the size of the respective TAD.
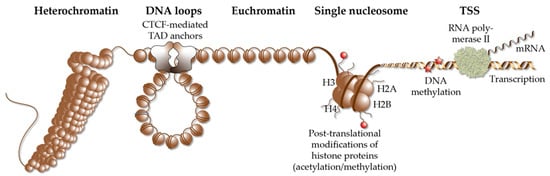
Figure 2. Elements of chromatin. Different elements of chromatin are shown, such as densely packed heterochromatin, DNA loops, such as TADs that are anchored by CTCF proteins, accessible euchromatin, the structure of a single nucleosome, chromatin modification via histone acetylation and methylation as well DNA methylation and a TSS, from which RNA polymerase II starts gene transcription into mRNA.
The distribution of eu- and heterochromatin (including their specific epigenetic markers) of a given cell is referred to as its epigenetic landscape or epigenome [35]. The epigenome is determined by patterns of DNA methylation, post-translational modification of histone tails and 3-dimensional chromatin organization [36]. It depends on the activity of chromatin-modifying enzymes, such as DNA methyltransferases (DNMTs), which add methyl groups to cytosines within genomic DNA, TET (Tet methylcytosine dioxygenase) proteins that initiate DNA methylation, or HATs, HDACs, lysine methyltransferases (KTMs) and KDMs, which add or remove acetyl and methyl groups to histones [37]. Histone acetylation is generally associated with transcriptional activation, but it is not important which exact amino acid is acetylated. In contrast, for histone methylation, the exact residue and its degree of methylation (mono-, di- or tri-methylation) is critical. Furthermore, the function of chromatin-remodeling enzymes is to shift or evict nucleosomes, in an ATP-dependent fashion. The projects ENCODE (www.encodeproject.org, accessed on 5 March 2022) [38] and Roadmap Epigenomics (www.roadmapepigenomics.org, accessed on 5 March 2022) [39] systematically assessed the epigenomes of more than 100 human cell lines, as well as primary cells, and serve as a reference for the epigenome of non-stimulated human tissues and cell types. However, in contrast to the static genome, the epigenome dynamically responds to intra- and extracellular signals, since chromatin modifying enzymes are often the endpoints of transduction cascades of peptide hormones, cytokines and growth factors [40]. Thus, the response of the epigenome to signals, such as 1,25(OH)2D3, is even more important than its ground state.
Nuclear hormones, such as 1,25(OH)2D3, affect the epigenome via direct interaction of their receptors with chromatin-modifying enzymes, as well as through up- or down-regulating the genes encoding for chromatin modifiers. In this way:
-
VDR initiates the demethylation of its binding sites via interaction with TET2 [42];
-
The binding of CTCF, to more than 1000 of its genomic sites, is modulated by 1,25(OH)2D3 [45];
-
The organization of some 400 TADs is dependent on 1,25(OH)2D3 [45], i.e., vitamin D affects the 3-dimensional chromatin structure.
Thus, there are multiple ways by which 1,25(OH)2D3 modulates the epigenome of its target tissues. Interestingly, some 1,25(OH)2D3-modulated chromatin loci, such as TSS regions, open only 2 h after ligand stimulation, while most sites take 24 h to reach maximal accessibility [41][43]. This suggests that many effects of vitamin D on the epigenome are secondary, i.e., they are mediated by genes and proteins that are primary vitamin D targets [46][47]. Nevertheless, the vitamin D-triggered effects on the epigenome facilitate the looping of VDR-bound enhancers, towards accessible TSS regions within the same TAD [48]. This assembly of enhancer and TSS regions enables the formation of a large protein complex, containing VDR, nuclear adaptor proteins, chromatin-modifying enzymes and RNA polymerase II, modulating gene transcription (Figure 1).
3. Vitamin D Target Genes
Vitamin D target genes are detected via a statistically significant change in their expression, within a given time frame (often 24 h), after ligand stimulation. Long-time known vitamin D target genes, such as BGLAP (bone gamma-carboxyglutamate protein, also called osteocalcin) [49] or CAMP (cathelicidin antimicrobial peptide) [50], were deduced based on the observation of the physiological effects of vitamin D, e.g., on calcium homeostasis or the defense against microbe infection, respectively. On the level of mRNA changes, vitamin D target genes were analyzed, initially, by single gene approaches, using northern blotting or quantitative PCR.
Time course analysis of vitamin D target genes allows one to classify them into rapidly responding (4–8 h) “primary” target genes and delayed-reacting “secondary” targets (Figure 3A). Primary target genes are directly regulated by 1,25(OH)2D3-activated VDR, as described in the model of vitamin D signaling; i.e., these genes need to have, within the same TAD, a VDR-binding enhancer. In contrast, secondary target genes may be regulated by transcription factors, co-factors or chromatin modifiers, which are encoded by primary vitamin D target genes (Figure 3A). Suitable proteins encoded by primary target genes are the transcription factors BCL6 (B-cell CLL/lymphoma 6), NFE2 (nuclear factor, erythroid 2), POU4F2 (POU class 4 homeobox 2) and ELF4 (E74-like factor 4) in THP-1 cells [46], as well as IRF5 (interferon regulatory factor 5), MAFF (MAF BZIP transcription factor F), MYCL (MYCL proto-oncogene, BHLH transcription factor), NFXL1 (nuclear transcription factor, X-box binding-like 1) and TFEC (transcription factor EC), as well as the transcriptional co-regulators MAMLD1 (mastermind-like domain-containing 1), PPARGC1B (PPARG coactivator 1 beta), SRA1 (steroid receptor RNA activator 1) and ZBTB46 (zinc finger and BTB domain-containing 46) in human PBMCs (peripheral blood mononuclear cells) [51]. In this way, secondary target genes do not have to carry a VDR-binding enhancer within their TADs. For example, although the time course study in human PBMCs used a strict statistical approach (threshold testing applying a fold change (FC) > 1.5 and 2), 662 vitamin D-responding genes were identified (FDR < 0.05), 179 of which are primary and 483 secondary targets [51]. An alternative classification of the same set of genes suggests that 293 of them are direct and 369 indirect targets of vitamin D (Figure 3B). Irrespective of the timing of their response to 1,25(OH)2D3, the expression change of direct targets is driven by VDR-bound enhancers, while indirect targets are primarily stabilized in their expression by 1,25(OH)2D3 and its receptor, against an up- or down-regulation by other transcription factors and/or their epigenetic effects.
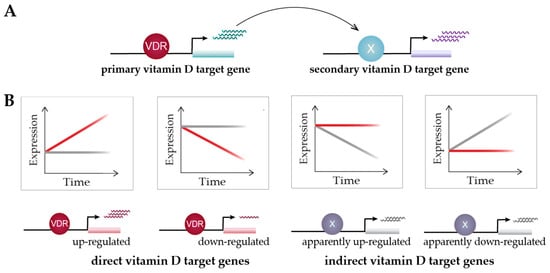
Figure 3. Classification of vitamin D target genes. Primary vitamin D target genes are directly regulated by VDR, while secondary vitamin D targets are controlled by transcriptional regulators that are encoded by primary targets (A). Time course analysis allows to differentiate vitamin D target genes in four different types based on cause and direction of expression change [51] (B). This suggests an alternative view on vitamin D signaling: 1,25(OH)2D3 either directly induces or reduces the expression of its target genes via VDR or prevents their expression change mediated by other factors. Red and grey lines indicate gene’s expression level in the presence or absence of 1,25(OH)2D3, respectively.
The model of vitamin D signaling illustrates the mechanisms of up-regulation of primary vitamin D target genes. However, the majority of vitamin D target genes are down-regulated, in particular, when cells are stimulated with 1,25(OH)2D3 for 24 h or longer. The down-regulation of a gene by vitamin D is only possible when the gene is first up-regulated by other transcription factors or epigenetic effects, mediated by chromatin modifiers. The most, likely mechanism of down-regulation of a gene is to block one or several of its up-regulating factors. Accordingly, the majority of down-regulated target genes could be classified as indirect targets, i.e., vitamin D counteracts their up-regulation rather than prominently down-regulating their expression [51]. For example, VDR antagonizes pro-inflammatory transcription factors, such as NFAT, AP1 and NFκB, in immune cells [52]. However, since each gene is up-regulated by an individual set of transcription factors and chromatin modifiers, there are also individual mechanisms of its down-regulation. Thus, there is no general model for describing the mechanism of the down-regulation of vitamin D target genes.
The expression of the majority of vitamin D target genes is less than 5-fold, up- or down-regulated (after a stimulation for 24 h with 1,25(OH)2D3); i.e., only a few genes respond with huge expression changes to vitamin D. For example, in THP-1 cells, the top five are the up-regulated genes CYP24A1, CAMP, TSPAN18 (tetraspanin 18), CD14 (CD14 molecule) and FBP1 (fructose-bisphosphatase 1), with an FC of expression ranging from 47 to 402 [43]. In human PBMCs, CYP24A1 and CAMP also have an FC of 409 and 158, respectively, the top up-regulated vitamin D target genes, but STEAP4 (STEAP4 metalloreductase), NRG1 (neuregulin 1) and CXCL10 (C-X-C motif chemokine ligand 10) show an FC of 471, 450 and 158, respectively, meaning comparable levels of down-regulation of their expression [51]. This prominent down-regulation of expression is more remarkable than the up-regulation, since it is far easier to increase a very low basal expression than a down-regulation of a highly expressed gene. Nevertheless, one should remember that these in vitro 1,25(OH)2D3 experiments are designed for maximal effects and do not reflect the reality of the endocrinology of vitamin D in vivo [53][54].
References
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263.
- Carlberg, C.; Molnár, F. Transcription factors. In Mechanisms of Gene Regulation, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 57–73.
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272.
- Carlberg, C.; Molnár, F. Switching genes on and off: The example of nuclear receptors. In Mechanisms of Gene Regulation, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 95–108.
- Molnár, F.; Peräkylä, M.; Carlberg, C. Vitamin D receptor agonists specifically modulate the volume of the ligand-binding pocket. J. Biol. Chem. 2006, 281, 10516–10526.
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892.
- Molnár, F. Structural considerations of vitamin D signaling. Front. Physiol. 2014, 5, 191.
- Tagami, T.; Lutz, W.H.; Kumar, R.; Jameson, J.L. The interaction of the vitamin D receptor with nuclear receptor corepressors and coactivators. Biochem. Biophys. Res. Commun. 1998, 253, 358–363.
- Polly, P.; Herdick, M.; Moehren, U.; Baniahmad, A.; Heinzel, T.; Carlberg, C. VDR-Alien: A novel, DNA-selective vitamin D3 receptor-corepressor partnership. FASEB J. 2000, 14, 1455–1463.
- Herdick, M.; Carlberg, C. Agonist-triggered modulation of the activated and silent state of the vitamin D3 receptor by interaction with co-repressors and co-activators. J. Mol. Biol. 2000, 304, 793–801.
- Rachez, C.; Lemon, B.D.; Suldan, Z.; Bromleigh, V.; Gamble, M.; Näär, A.M.; Erdjument-Bromage, H.; Tempst, P.; Freedman, L.P. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 1999, 398, 824–828.
- Belorusova, A.Y.; Bourguet, M.; Hessmann, S.; Chalhoub, S.; Kieffer, B.; Cianferani, S.; Rochel, N. Molecular determinants of MED1 interaction with the DNA bound VDR-RXR heterodimer. Nucleic Acids Res. 2020, 48, 11199–11213.
- Yuan, C.-X.; Ito, M.; Fondell, J.D.; Fu, Z.-Y.; Roeder, R.G. The TRAP220 component of a thyroid hormone receptor-associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc. Natl. Acad. Sci. USA 1998, 95, 7939–7944.
- Pereira, F.; Barbachano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Munoz, A.; Larriba, M.J. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665.
- Battaglia, S.; Karasik, E.; Gillard, B.; Williams, J.; Winchester, T.; Moser, M.T.; Smiraglia, D.J.; Foster, B.A. LSD1 dual function in mediating epigenetic corruption of the vitamin D signaling in prostate cancer. Clin. Epigenet. 2017, 9, 82.
- Wei, Z.; Yoshihara, E.; He, N.; Hah, N.; Fan, W.; Pinto, A.F.M.; Huddy, T.; Wang, Y.; Ross, B.; Estepa, G.; et al. Vitamin D switches BAF complexes to protect beta cells. Cell 2018, 173, 1135–1149.e15.
- Cui, X.; Pertile, R.; Eyles, D.W. The vitamin D receptor (VDR) binds to the nuclear matrix via its hinge domain: A potential mechanism for the reduction in VDR mediated transcription in mitotic cells. Mol. Cell. Endocrinol. 2018, 472, 18–25.
- Schräder, M.; Bendik, I.; Becker-Andre, M.; Carlberg, C. Interaction between retinoic acid and vitamin D signaling pathways. J. Biol. Chem. 1993, 268, 17830–17836.
- Schräder, M.; Müller, K.M.; Carlberg, C. Specificity and flexibility of vitamin D signaling.: Modulation of the activation of natural vitamin D response elements by thyroid hormone. J. Biol. Chem. 1994, 269, 5501–5504.
- Schräder, M.; Müller, K.M.; Nayeri, S.; Kahlen, J.P.; Carlberg, C. VDR-T3R receptor heterodimer polarity directs ligand sensitivity of transactivation. Nature 1994, 370, 382–386.
- Carlberg, C.; Molnár, F. Vitamin D receptor signaling and its therapeutic implications: Genome-wide and structural view. Can. J. Physiol. Pharmacol. 2015, 93, 311–318.
- Tuoresmäki, P.; Väisänen, S.; Neme, A.; Heikkinen, S.; Carlberg, C. Patterns of genome-wide VDR locations. PLoS ONE 2014, 9, e96105.
- Seuter, S.; Neme, A.; Carlberg, C. Epigenomic PU.1-VDR crosstalk modulates vitamin D signaling. Biochim. Biophys. Acta 2017, 1860, 405–415.
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011, 144, 296–309.
- Nurminen, V.; Neme, A.; Seuter, S.; Carlberg, C. Modulation of vitamin D signaling by the pioneer factor CEBPA. Biochim. Biophys. Acta 2019, 1862, 96–106.
- Seuter, S.; Neme, A.; Carlberg, C. ETS transcription factor family member GABPA contributes to vitamin D receptor target gene regulation. J. Steroid Biochem. Mol. Biol. 2018, 177, 46–52.
- Warwick, T.; Schulz, M.H.; Gunther, S.; Gilsbach, R.; Neme, A.; Carlberg, C.; Brandes, R.P.; Seuter, S. A hierarchical regulatory network analysis of the vitamin D induced transcriptome reveals novel regulators and complete VDR dependency in monocytes. Sci. Rep. 2021, 11, 6518.
- Meyer, M.B.; Benkusky, N.A.; Sen, B.; Rubin, J.; Pike, J.W. Epigenetic plasticity drives adipogenic and osteogenic differentiation of marrow-derived mesenchymal stem cells. J. Biol. Chem. 2016, 291, 17829–17847.
- Chauss, D.; Freiwald, T.; McGregor, R.; Yan, B.; Wang, L.; Nova-Lamperti, E.; Kumar, D.; Zhang, Z.; Teague, H.; West, E.E.; et al. Autocrine vitamin D signaling switches off pro-inflammatory programs of TH1 cells. Nat. Immunol. 2022, 23, 62–74.
- Ali, T.; Renkawitz, R.; Bartkuhn, M. Insulators and domains of gene expression. Curr. Opin. Genet. Dev. 2016, 37, 17–26.
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380.
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211.
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891.
- Felsenfeld, G.; Burgess-Beusse, B.; Farrell, C.; Gaszner, M.; Ghirlando, R.; Huang, S.; Jin, C.; Litt, M.; Magdinier, F.; Mutskov, V.; et al. Chromatin boundaries and chromatin domains. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 245–250.
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398.
- Carlberg, C.; Molnár, F. The epigenome. In Mechanisms of Gene Regulation, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016.
- Carlberg, C.; Molnár, F. Chromatin modifiers. In Mechanisms of Gene Regulation, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016.
- ENCODE-Project-Consortium; Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74.
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330.
- Carlberg, C.; Molnár, F. Human Epigenetics: How Science Works; Springer: New York, NY, USA, 2019.
- Nurminen, V.; Neme, A.; Seuter, S.; Carlberg, C. The impact of the vitamin D-modulated epigenome on VDR target gene regulation. Biochim. Biophys. Acta 2018, 1861, 697–705.
- Catala-Moll, F.; Ferrete-Bonastre, A.G.; Godoy-Tena, G.; Morante-Palacios, O.; Ciudad, L.; Barbera, L.; Fondelli, F.; Martinez-Caceres, E.M.; Rodriguez-Ubreva, J.; Li, T.; et al. Vitamin D receptor, STAT3, and TET2 cooperate to establish tolerogenesis. Cell Rep. 2022, 38, 110244.
- Seuter, S.; Neme, A.; Carlberg, C. Epigenome-wide effects of vitamin D and their impact on the transcriptome of human monocytes involve CTCF. Nucleic Acids Res. 2016, 44, 4090–4104.
- Seuter, S.; Pehkonen, P.; Heikkinen, S.; Carlberg, C. Dynamics of 1α,25-dihydroxyvitamin D-dependent chromatin accessibility of early vitamin D receptor target genes. Biochim. Biophys. Acta 2013, 1829, 1266–1275.
- Neme, A.; Seuter, S.; Carlberg, C. Vitamin D-dependent chromatin association of CTCF in human monocytes. Biochim. Biophys. Acta 2016, 1859, 1380–1388.
- Nurminen, V.; Neme, A.; Ryynanen, J.; Heikkinen, S.; Seuter, S.; Carlberg, C. The transcriptional regulator BCL6 participates in the secondary gene regulatory response to vitamin D. Biochim. Biophys. Acta 2015, 1849, 300–308.
- Nurminen, V.; Seuter, S.; Carlberg, C. Primary vitamin D target genes of human monocytes. Front. Physiol. 2019, 10, 194.
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136.
- Lian, J.B.; Glimcher, M.J.; Roufosse, A.H.; Hauschka, P.V.; Gallop, P.M.; Cohen-Solal, L.; Reit, B. Alterations of the gamma-carboxyglutamic acid and osteocalcin concentrations in vitamin D-deficient chick bone. J. Biol. Chem. 1982, 257, 4999–5003.
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077.
- Hanel, A.; Carlberg, C. Time-resolved gene expression analysis monitors the regulation of inflammatory mediators and attenuation of adaptive immune response by vitamin D. Int. J. Mol. Sci. 2022, 23, 911.
- Zeitelhofer, M.; Adzemovic, M.Z.; Gomez-Cabrero, D.; Bergman, P.; Hochmeister, S.; N’Diaye, M.; Paulson, A.; Ruhrmann, S.; Almgren, M.; Tegner, J.N.; et al. Functional genomics analysis of vitamin D effects on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2017, 114, E1678–E1687.
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689S–1696S.
- Norman, A.W. From vitamin D to hormone D: Fundamentals of the vitamin D endocrine system essential for good health. Am. J. Clin. Nutr. 2008, 88, 491S–499S.
More
Information
Subjects:
Nutrition & Dietetics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revision:
1 time
(View History)
Update Date:
12 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No