 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yi-Qing Yang | -- | 2097 | 2022-04-09 16:39:30 | | | |
| 2 | Jason Zhu | -1 word(s) | 2096 | 2022-04-11 05:11:38 | | |
Video Upload Options
Atrial fibrillation (AF) represents the most common type of clinical cardiac arrhythmia worldwide and contributes to substantial morbidity, mortality and socioeconomic burden. Aggregating evidence highlights the strong genetic basis of AF. In addition to chromosomal abnormalities, pathogenic mutations in over 50 genes have been causally linked to AF, of which the majority encode ion channels, cardiac structural proteins, transcription factors and gap junction channels. In the heart, gap junctions comprised of connexins (Cxs) form intercellular pathways responsible for electrical coupling and rapid coordinated action potential propagation between adjacent cardiomyocytes. Among the 21 isoforms of connexins already identified in the mammal genomes, 5 isoforms (Cx37, Cx40, Cx43, Cx45 and Cx46) are expressed in human heart. Abnormal electrical coupling between cardiomyocytes caused by structural remodeling of gap junction channels (alterations in connexin distribution and protein levels) has been associated with enhanced susceptibility to AF and recent studies have revealed multiple causative mutations or polymorphisms in 4 isoforms of connexins predisposing to AF.
1. Introduction
2. Structure and Function of Gap Junctions

3. Subtypes of Cardiac Connexins
4. Changes of Gap Junctions/Connexins in the Pathogenesis of AF
4.1. Gap Junction Remodeling
4.2. Abundance and Distribution of Connexins Associated with AF
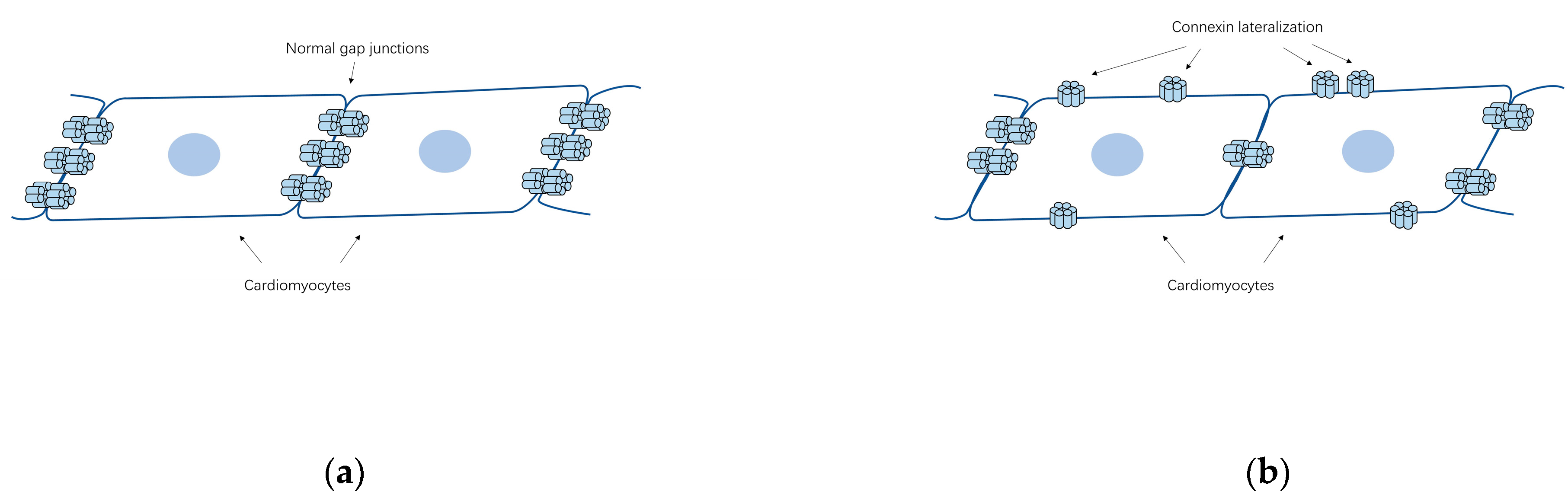
4.3. Changes in Atrial Connexin Expression Regulated by Transcription Factors
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528.
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375.
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867.
- Fuster, V.; Ryden, L.E.; Cannom, D.S.; Crijns, H.J.; Curtis, A.B.; Ellenbogen, K.A.; Halperin, J.L.; Kay, G.N.; Le Huezey, J.Y.; Lowe, J.E.; et al. American College of Cardiology Foundation/American Heart Association Task Force: 2011 ACCF/AHA/HRS focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011, 123, e269–e367.
- Zimetbaum, P. Atrial Fibrillation. Ann. Intern. Med. 2017, 166, ITC33–ITC48.
- Saffitz, J.E. Connexins, conduction, and atrial fibrillation. N. Engl. J. Med. 2006, 354, 2712–2714.
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014, 130, e199–e267.
- Campuzano, O.; Perez-Serra, A.; Iglesias, A.; Brugada, R. Genetic basis of atrial fibrillation. Genes Dis. 2016, 3, 257–262.
- Fox, C.S.; Parise, H.; D’Agostino, R.B.; Lloyd-Jones, D.M.; Vasan, R.S.; Wang, T.J.; Levy, D.; Wolf, P.A.; Benjamin, E.J. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. J. Am. Med. Assoc. 2004, 291, 2851–2855.
- Guo, X.J.; Qiu, X.B.; Wang, J.; Guo, Y.H.; Yang, C.X.; Li, L.; Gao, R.F.; Ke, Z.P.; Di, R.M.; Sun, Y.M.; et al. PRRX1 Loss-of-Function Mutations Underlying Familial Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e023517.
- Severs, N.J.; Dupont, E.; Thomas, N.; Kaba, R.; Rothery, S.; Jain, R.; Sharpey, K.; Fry, C.H. Alterations in cardiac connexin expression in cardiomyopathies. Adv. Cardiol. 2006, 42, 228–242.
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576.
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211.
- Chaldoupi, S.M.; Loh, P.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. The role of connexin40 in atrial fibrillation. Cardiovasc. Res. 2009, 84, 15–23.
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035.
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 5–8.
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19.
- Duffy, H.S.; Wit, A.L. Is there a role for remodeled connexins in AF? No simple answers. J. Mol. Cell. Cardiol. 2008, 44, 4–13.
- Ortega, A.; Tarazon, E.; Gil-Cayuela, C.; Garcia-Manzanares, M.; Martinez-Dolz, L.; Lago, F.; Gonzalez-Juanatey, J.R.; Cinca, J.; Jorge, E.; Portoles, M.; et al. Intercalated disc in failing hearts from patients with dilated cardiomyopathy: Its role in the depressed left ventricular function. PLoS ONE 2017, 12, e0185062.
- Desplantez, T. Cardiac Cx43, Cx40 and Cx45 co-assembling: Involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell. Biol. 2017, 18, 3.
- van der Velden, H.M.; Jongsma, H.J. Cardiac gap junctions and connexins: Their role in atrial fibrillation and potential as therapeutic targets. Cardiovasc. Res. 2002, 54, 270–279.
- Severs, N.J.; Coppen, S.R.; Dupont, E.; Yeh, H.I.; Ko, Y.S.; Matsushita, T. Gap junction alterations in human cardiac disease. Cardiovasc. Res. 2004, 62, 368–377.
- Cheng, S.H.; Shakespeare, T.; Mui, R.; White, T.W.; Valdimarsson, G. Connexin 48.5 is required for normal cardiovascular function and lens development in zebrafish embryos. J. Biol. Chem. 2004, 279, 36993–37003.
- Reaume, A.G.; Desousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.G.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac Malformation in Neonatal Mice Lacking Connexin43. Science 1995, 267, 1831–1834.
- Gu, H.; Smith, F.C.; Taffet, S.M.; Delmar, M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003, 93, 201–206.
- Seki, A.; Ishikawa, T.; Daumy, X.; Mishima, H.; Barc, J.; Sasaki, R.; Nishii, K.; Saito, K.; Urano, M.; Ohno, S.; et al. Progressive Atrial Conduction Defects Associated With Bone Malformation Caused by a Connexin-45 Mutation. J. Am. Coll. Cardiol. 2017, 70, 358–370.
- Munger, S.J.; Kanady, J.D.; Simon, A.M. Absence of venous valves in mice lacking Connexin37. Dev. Biol. 2013, 373, 338–348.
- Kanady, J.D.; Munger, S.J.; Witte, M.H.; Simon, A.M. Combining Foxc2 and Connexin37 deletions in mice leads to severe defects in lymphatic vascular growth and remodeling. Dev. Biol. 2015, 405, 33–46.
- Martins-Marques, T.; Catarino, S.; Goncalves, A.; Miranda-Silva, D.; Goncalves, L.; Antunes, P.; Coutinho, G.; Moreira, A.L.; Pires, I.F.; Girao, H. EHD1 Modulates Cx43 Gap Junction Remodeling Associated With Cardiac Diseases. Circ. Res. 2020, 126, E97–E113.
- Tribulova, N.; Knezl, V.; Okruhlicova, L.; Slezak, J. Myocardial Gap Junctions: Targets for Novel Approaches in the Prevention of Life-Threatening Cardiac Arrhythmias. Physiol Res 2008, 57, S1–S13.
- Spach, M.S.; Starmer, C.F. Altering the Topology of Gap-Junctions—A Major Therapeutic Target for Atrial-Fibrillation. Cardiovasc. Res. 1995, 30, 337–344.
- Kato, T.; Iwasaki, Y.K.; Nattel, S. Connexins and Atrial Fibrillation Filling in the Gaps. Circulation 2012, 125, 203–206.
- Wetzel, U.; Boldt, A.; Lauschke, J.; Weigl, J.; Schirdewahn, P.; Dorszewski, A.; Doll, N.; Hindricks, G.; Dhein, S.; Kottkamp, H. Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 2005, 91, 166–170.
- Polontchouk, L.; Haefliger, J.A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.R.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891.
- Kanagaratnam, P.; Cherian, A.; Stanbridge, R.D.L.; Glenville, B.; Severs, N.J.; Peters, N.S. Relationship between connexins and atrial activation during human atrial fibrillation. J. Cardiovasc. Electr. 2004, 15, 206–213.
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379.
- Dhein, S.; Rothe, S.; Busch, A.; Rojas Gomez, D.M.; Boldt, A.; Reutemann, A.; Seidel, T.; Salameh, A.; Pfannmuller, B.; Rastan, A.; et al. Effects of metoprolol therapy on cardiac gap junction remodelling and conduction in human chronic atrial fibrillation. Br. J. Pharmacol. 2011, 164, 607–616.
- Rucker-Martin, C.; Milliez, P.; Tan, S.; Decrouy, X.; Recouvreur, M.; Vranckx, R.; Delcayre, C.; Renaud, J.F.; Dunia, I.; Segretain, D.; et al. Chronic hemodynamic overload of the atria is an important factor for gap junction remodeling in human and rat hearts. Cardiovasc. Res. 2006, 72, 69–79.
- Fakuade, F.E.; Tomsits, P.; Voigt, N. Connexin hemichannels in atrial fibrillation: Orphaned and irrelevant? Cardiovasc. Res. 2021, 117, 4–6.
- Dhein, S.; Salameh, A. Remodeling of Cardiac Gap Junctional Cell-Cell Coupling. Cells 2021, 10, 2422.
- Jungk, L.; Franke, H.; Salameh, A.; Dhein, S. Golgi Fragmentation in Human Patients with Chronic Atrial Fibrillation: A New Aspect of Remodeling. Thorac. Cardiov. Surg. 2019, 67, 98–106.
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133.
- Ma, J.F.; Yang, F.; Mahida, S.N.; Zhao, L.; Chen, X.; Zhang, M.L.; Sun, Z.; Yao, Y.; Zhang, Y.X.; Zheng, G.Y.; et al. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc. Res. 2016, 109, 442–450.
- Mechakra, A.; Footz, T.; Walter, M.; Aranega, A.; Hernandez-Torres, F.; Morel, E.; Millat, G.; Yang, Y.Q.; Chahine, M.; Chevalier, P.; et al. A Novel PITX2c Gain-of-Function Mutation, p.Met207Val, in Patients With Familial Atrial Fibrillation. Am. J. Cardiol. 2019, 123, 787–793.
- Yan, J.; Kong, W.; Zhang, Q.; Beyer, E.C.; Walcott, G.; Fast, V.G.; Ai, X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc. Res. 2013, 97, 589–597.

