 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Weifang Liu | -- | 3112 | 2022-04-06 19:20:30 | | | |
| 2 | Conner Chen | -117 word(s) | 2995 | 2022-04-07 05:10:12 | | |
Video Upload Options
Human brain, a central organ of the human nervous system, is a highly complex organ that regulates many essential processes including cognition, memory, emotion, vision, breathing, motor skills, and experiences of surroundings. As the most complex organ in the human body, the brain manifests its complexity in various aspects. Underneath the cerebral cortex, there are many indispensable structures encompassing the thalamus, the epithalamus, the striatum, the pineal gland, the pituitary gland, the hypothalamus, the subthalamus, the substantia nigra, as well as the limbic structures, including the amygdala and the hippocampus. A number of studies, particularly through examining gene expression and epigenetic profiles from various regions of the brain, have identified the most associated regions for different brain-related disorders.
1. Folding Principles of the Human Genome
2. Human Brain Genome Organization and Its Relevance to Neuropsychiatric Disorders
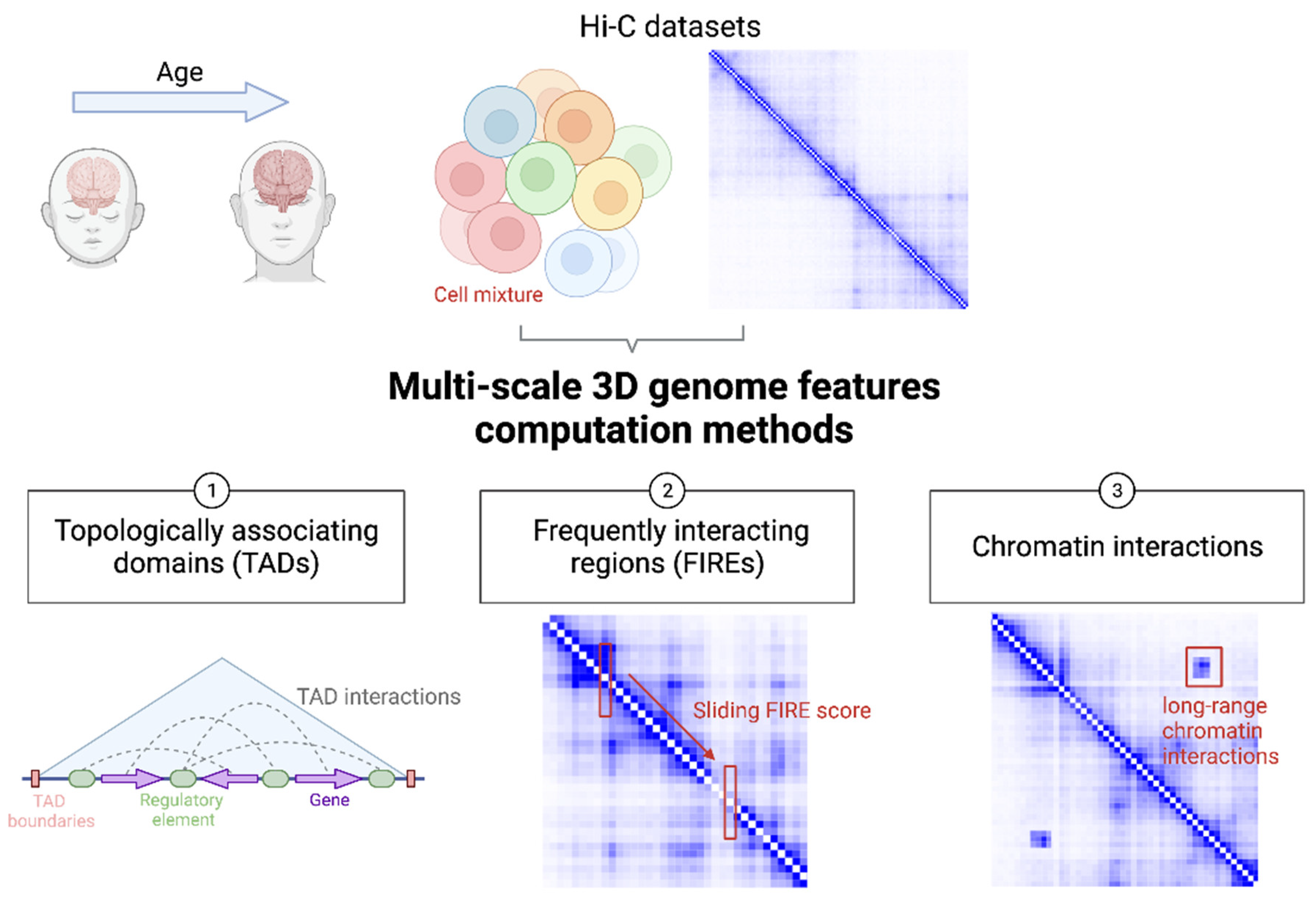
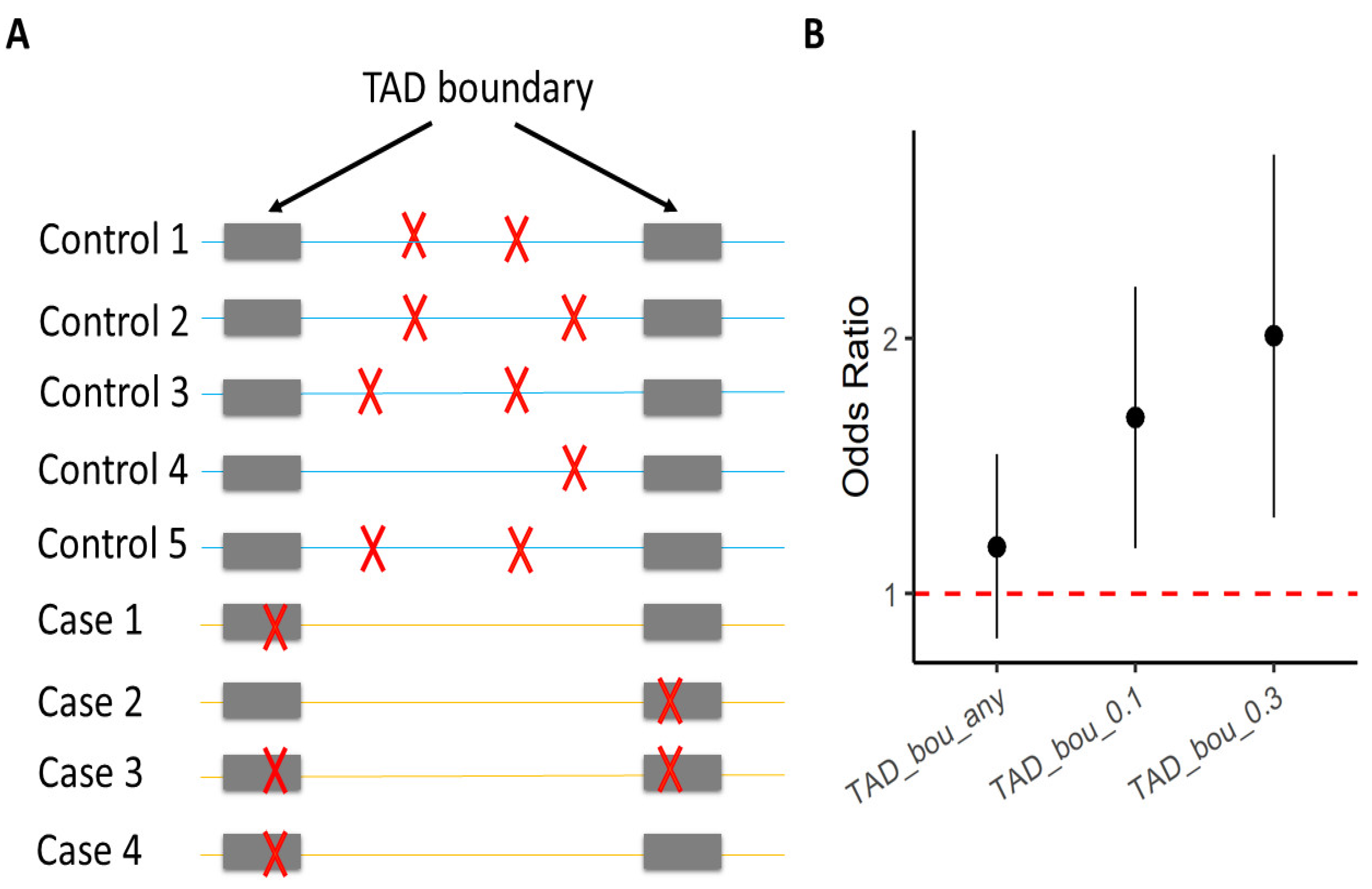
3. TAD
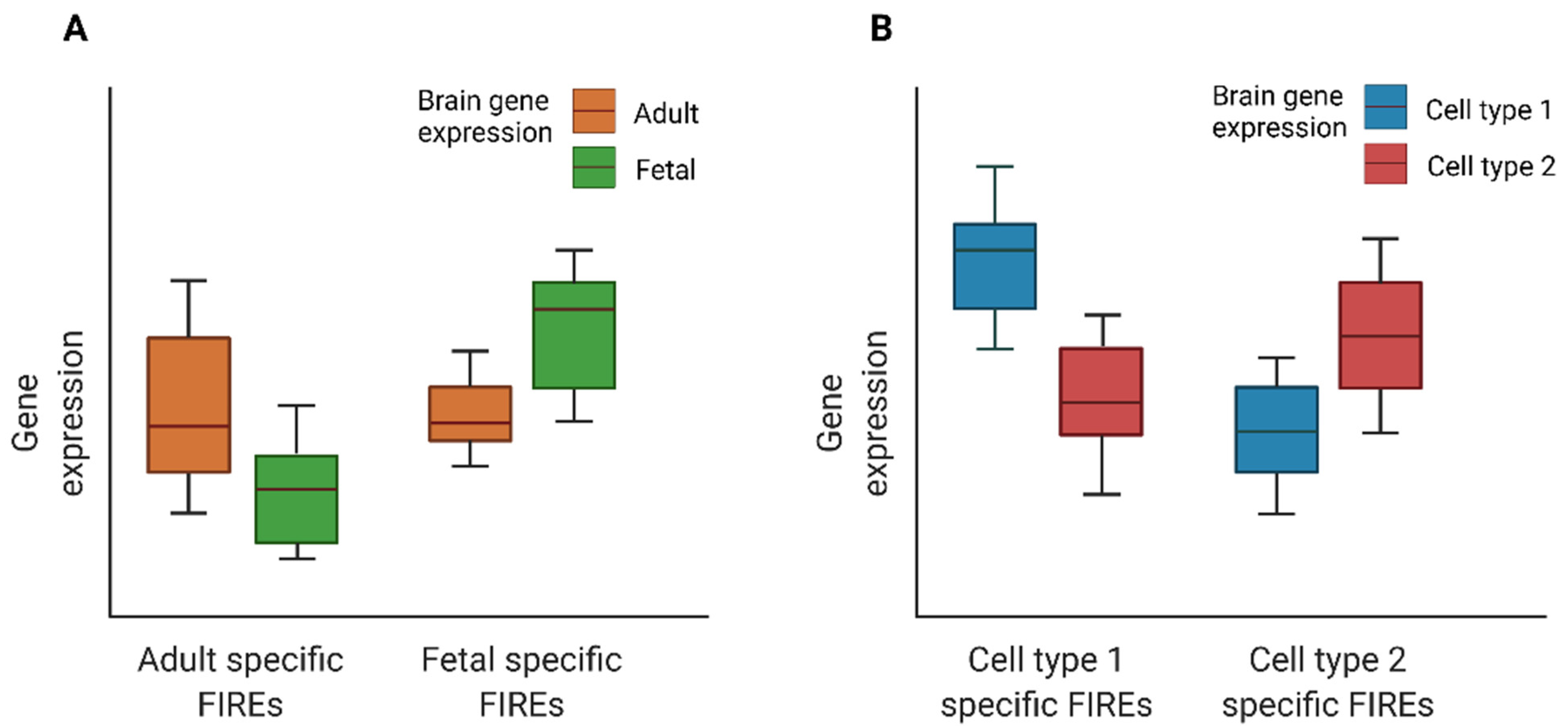
4. FIRE
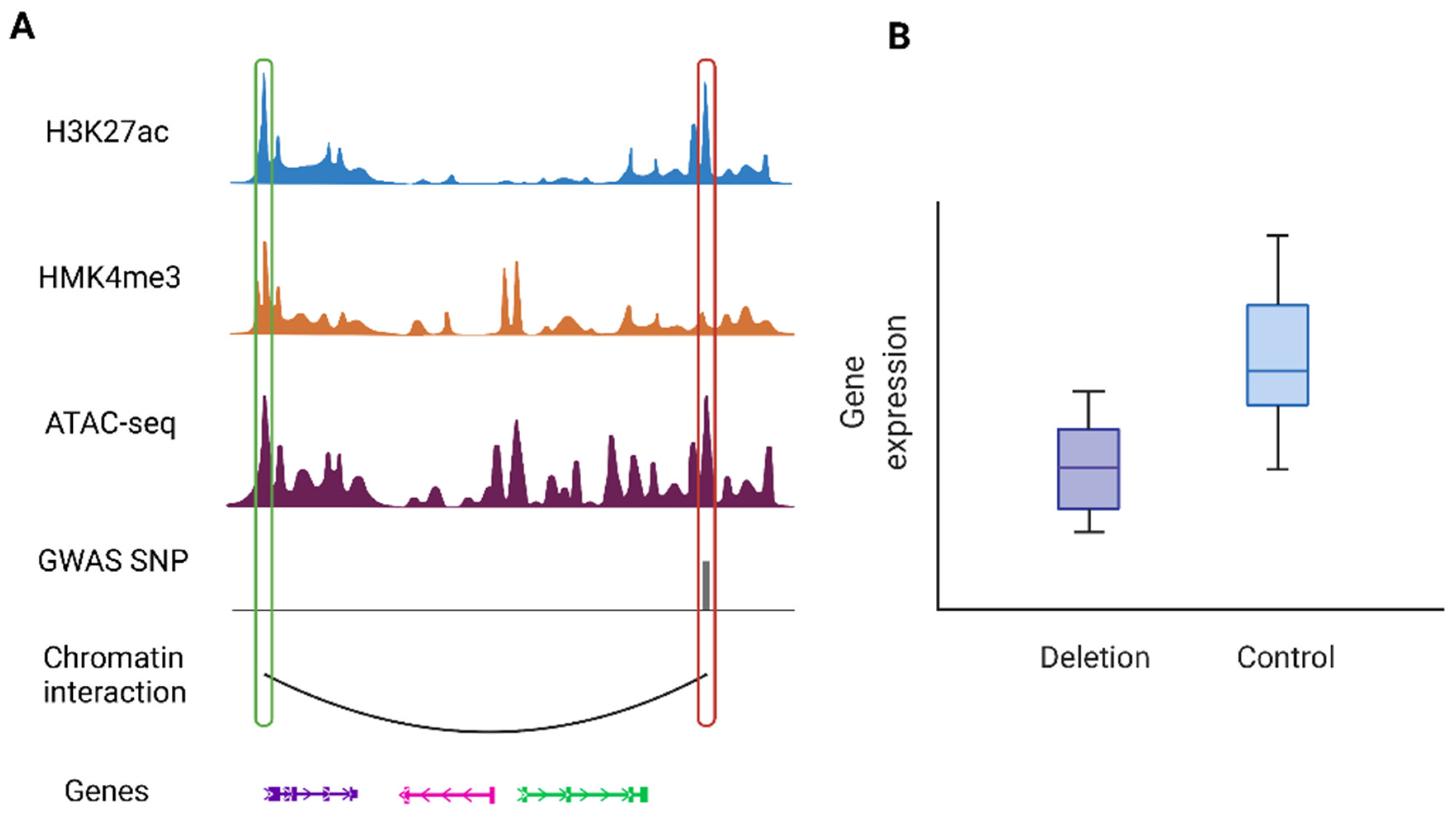
5. Chromatin Interactions
References
- Schmitt, A.D.; Hu, M.; Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016, 17, 743–755.
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678.
- Li, Y.; Hu, M.; Shen, Y. Gene regulation in the 3D genome. Hum. Mol. Genet. 2018, 27, R228–R233.
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800.
- Jerkovic, I.; Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 2021, 22, 511–528.
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550.
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737.
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293.
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680.
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572.e24.
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016, 17, 2042–2059.
- Cremer, T.; Cremer, M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889.
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380.
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385.
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455.
- Lagler, T.M.; Abnousi, A.; Hu, M.; Yang, Y.; Li, Y. HiC-ACT: Improved detection of chromatin interactions from Hi-C data via aggregated Cauchy test. Am. J. Hum. Genet. 2021, 108, 257–268.
- Martin, J.S.; Xu, Z.; Reiner, A.P.; Mohlke, K.L.; Sullivan, P.; Ren, B.; Hu, M.; Li, Y. HUGIn: Hi-C Unifying Genomic Interrogator. Bioinformatics 2017, 33, 3793–3795.
- Stadhouders, R.; Filion, G.J.; Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 2019, 569, 345–354.
- Krijger, P.H.L.; de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016, 17, 771–782.
- Schulz, J.M.; Al-Khazraji, B.K.; Shoemaker, J.K. Sodium nitroglycerin induces middle cerebral artery vasodilatation in young, healthy adults. Exp. Physiol. 2018, 103, 1047–1055.
- Simpkins, C.A.; Simpkins, A.M. Neuroscience for Clinicians: Evidence, Models, and Practice; Springer: Berlin/Heidelberg, Germany, 2012; p. 143. ISBN 978-1-4614-4842-6.
- Bassett, D.S.; Gazzaniga, M.S. Understanding complexity in the human brain. Trends Cogn. Sci. (Regul. Ed.) 2011, 15, 200–209.
- O’Leary, D.H.; Polak, J.F.; Kronmal, R.A.; Manolio, T.A.; Burke, G.L.; Wolfson, S.K. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N. Engl. J. Med. 1999, 340, 14–22.
- Jevtic, S.; Sengar, A.S.; Salter, M.W.; McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res. Rev. 2017, 40, 84–94.
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430.
- Bryois, J.; Skene, N.G.; Hansen, T.F.; Kogelman, L.J.A.; Watson, H.J.; Liu, Z.; Eating Disorders Working Group of the Psychiatric Genomics Consortium; International Headache Genetics Consortium; 23andMe Research Team; Brueggeman, L.; et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 2020, 52, 482–493.
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The translational significance of the neurovascular unit. J. Biol. Chem. 2017, 292, 762–770.
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA 2015, 112, 7285–7290.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337.
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677.
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546.
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019, 51, 1252–1262.
- Nott, A.; Holtman, I.R.; Coufal, N.G.; Schlachetzki, J.C.M.; Yu, M.; Hu, R.; Han, C.Z.; Pena, M.; Xiao, J.; Wu, Y.; et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019, 366, 1134–1139.
- Song, M.; Pebworth, M.-P.; Yang, X.; Abnousi, A.; Fan, C.; Wen, J.; Rosen, J.D.; Choudhary, M.N.K.; Cui, X.; Jones, I.R.; et al. Cell-type-specific 3D epigenomes in the developing human cortex. Nature 2020, 587, 644–649.
- Hu, B.; Won, H.; Mah, W.; Park, R.B.; Kassim, B.; Spiess, K.; Kozlenkov, A.; Crowley, C.A.; Pochareddy, S.; PsychENCODE Consortium; et al. Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021, 12, 3968.
- Li, M.; Santpere, G.; Imamura Kawasawa, Y.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615.
- Crowley, C.; Yang, Y.; Qiu, Y.; Hu, B.; Abnousi, A.; Lipiński, J.; Plewczyński, D.; Wu, D.; Won, H.; Ren, B.; et al. FIREcaller: Detecting frequently interacting regions from Hi-C data. Comput. Struct. Biotechnol. J. 2021, 19, 355–362.
- Giusti-Rodriguez, P.; Lu, L.; Yang, Y.; Crowley, C.A.; Liu, X.; Juric, I.; Martin, J.S.; Abnousi, A.; Allred, S.C.; Ancalade, N.; et al. Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. BioRxiv 2018.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427.
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Walters, J.T.; O’Donovan, M.C. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv 2020.
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. medRxiv 2020.
- Halvorsen, M.; Huh, R.; Oskolkov, N.; Wen, J.; Netotea, S.; Giusti-Rodriguez, P.; Karlsson, R.; Bryois, J.; Nystedt, B.; Ameur, A.; et al. Increased burden of ultra-rare structural variants localizing to boundaries of topologically associated domains in schizophrenia. Nat. Commun. 2020, 11, 1842.
- Gazal, S.; Finucane, H.K.; Furlotte, N.A.; Loh, P.-R.; Palamara, P.F.; Liu, X.; Schoech, A.; Bulik-Sullivan, B.; Neale, B.M.; Gusev, A.; et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 2017, 49, 1421–1427.
- Wainschtein, P.; Jain, D.P.; Yengo, L.; Zheng, Z.; TOPMed Anthropometry Working Group; Trans-Omics for Precision Medicine Consortium; Cupples, L.A.; Shadyab, A.H.; McKnight, B.; Shoemaker, B.M.; et al. Recovery of trait heritability from whole genome sequence data. BioRxiv 2019.
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025.
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.-A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45.
- Yu, M.; Abnousi, A.; Zhang, Y.; Li, G.; Lee, L.; Chen, Z.; Fang, R.; Lagler, T.M.; Yang, Y.; Wen, J.; et al. SnapHiC: A computational pipeline to identify chromatin loops from single-cell Hi-C data. Nat. Methods 2021, 18, 1056–1059.
- Sun, Q.; Crowley, C.A.; Huang, L.; Wen, J.; Chen, J.; Bao, E.L.; Auer, P.L.; Lettre, G.; Reiner, A.P.; Sankaran, V.G.; et al. From GWAS variant to function: A study of ∼148,000 variants for blood cell traits. HGG Adv. 2022, 3, 100063.
- Zhao, B.; Shan, Y.; Yang, Y.; Yu, Z.; Li, T.; Wang, X.; Luo, T.; Zhu, Z.; Sullivan, P.; Zhao, H.; et al. Transcriptome-wide association analysis of brain structures yields insights into pleiotropy with complex neuropsychiatric traits. Nat. Commun. 2021, 12, 2878.
- Walters, R.K.; Polimanti, R.; Johnson, E.C.; McClintick, J.N.; Adams, M.J.; Adkins, A.E.; Aliev, F.; Bacanu, S.-A.; Batzler, A.; Bertelsen, S.; et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 2018, 21, 1656–1669.
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444.
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681.
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527.
- Zhou, X.; Chen, Y.; Mok, K.Y.; Kwok, T.C.Y.; Mok, V.C.T.; Guo, Q.; Ip, F.C.; Chen, Y.; Mullapudi, N.; Alzheimer’s Disease Neuroimaging Initiative; et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat. Commun. 2019, 10, 3310.
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451.
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Serra Mari, R.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117.
- Johnston, M.J.; Nikolic, A.; Ninkovic, N.; Guilhamon, P.; Cavalli, F.M.G.; Seaman, S.; Zemp, F.J.; Lee, J.; Abdelkareem, A.; Ellestad, K.; et al. High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome Res. 2019, 29, 1211–1222.
- Wang, J.; Huang, T.Y.-T.; Hou, Y.; Bartom, E.; Lu, X.; Shilatifard, A.; Yue, F.; Saratsis, A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 2021, 7, eabg4126.

