1. Folding Principles of the Human Genome
The human genome consists of approximately 3 billion nucleotides, which can form a ~2-meter-long polymer if stretched in one-dimensional (1D) space. However, the average diameter of the nucleus in human cells is ~6 μm. The five orders of magnitude compaction from 1D space to 3D space results in highly complex chromatin spatial organization. How chromatin folds in 3D space have fascinated scientists during the last few decades. Deep understanding of the principles of chromatin folding holds great promise to reveal the structural basis of gene regulation and genome function
[1][2][3][4][5][6][7].
To achieve this goal, genome-wide experimental assays are essential to accurately characterize the 3D genome in the nucleus. Harnessing the power of next generation sequencing technologies, high-throughput chromatin conformation capture (Hi-C)
[8] has been widely applied to cultured cell lines, purified cell types, and complex tissues
[9][10][11], and has revealed 3D genome features at a cascade of resolutions. Specifically, at chromosome resolution, different chromosomes occupy distinct locations in the nucleus, termed as chromosome territories (CTs)
[12], where transcriptionally active regions are near the nuclear center, while transcriptionally inactive regions are near the nuclear periphery and tend to be associated with the nuclear lamina. Zooming in, each chromosome consists of mega-base (Mb) resolution A/B compartments
[8] and sub-compartments
[9], with more frequent interactions within the same type of compartment, and infrequent interactions between the two different types of compartments. The compartments can be further divided into topologically associating domains (TADs)
[13][14], which are typically several hundred kilobase to ~1 Mb in size. TADs function as the basic building block of the 3D genome, dictating the majority of chromatin interactions to be within the same TAD. Moving to finer tens of kilobase resolution, it has identified frequently interacting regions (FIREs) that mark regions of the genome and have a high level of interactions with their neighboring regions
[11]. Finally, at the finest kilobase resolution, two types of chromatin loops have been discovered, mostly inside of TADs. One type is structural loops, which are mediated by the convergent CTCF motif pairs and largely conserved among different cell types
[9]. The other type is functional loops, which are formed by enhancer–promoter interactions and exhibit high cell-type specificity
[15][16][17]. Extensive studies have demonstrated that these 3D genome features are closely related to transcriptional regulation mechanisms, the determination of cell identity, and organism-level health and disease outcomes
[18][19][20].
2. Human Brain Genome Organization and Its Relevance to Neuropsychiatric Disorders
Human brain, a central organ of the human nervous system, is a highly complex organ that regulates many essential processes including cognition, memory, emotion, vision, breathing, motor skills, and experiences of surroundings
[21]. As the most complex organ in the human body, the brain manifests its complexity in various aspects. First, the human brain exhibits complicated spatial organization
[22]. Specifically, it has the six-layered cerebral cortex, shared with other mammals, but notably larger in size. Underneath the cerebral cortex, there are many indispensable structures encompassing the thalamus, the epithalamus, the striatum, the pineal gland, the pituitary gland, the hypothalamus, the subthalamus, the substantia nigra, as well as the limbic structures, including the amygdala and the hippocampus. A number of studies
[23][24][25][26][27], particularly through examining gene expression and epigenetic profiles from various regions of the brain, have identified the most associated regions for different brain-related disorders. For example, schizophrenia (SCZ), intelligence, educational attainment, neuroticism, and major depressive disorder (MDD) have been found to be most significantly associated with the cortical regions; Parkinson’s disease was found to be most strongly associated with the expected substantia nigra; while Alzheimer’s disease (AD) shows consistent association with tissues playing prominent immune-related roles from multiple studies
[25][26][27].
Besides the numerous anatomical structures aforementioned, the human brain consists of diverse cell types. Specifically, there are two major categories of cell types in the brain, namely, neuronal cells and glial cells, as well as other cell types including vascular cells (such as pericytes) and endothelial cells
[28][29][30]. The glial cells can be further divided into astrocytes, oligodendrocytes, and microglia
[31]. Neuronal cells encompass an extraordinary diversity and can be further divided into dozens of subtypes under the two major cell subtypes: excitatory neurons and inhibitory neurons
[29][32]. As gene regulation varies substantially across cell types, and relevant cell types differ for different diseases and traits, it is important to study chromatin spatial organization across diverse cell types and understand gene regulation mechanisms in a cell-type-specific manner. Some recent efforts have been made, including the interrogation of neuronal cell types derived from induced pluripotent stem cells, and also of primary cells obtained through cell sorting including different types of neurons, astrocytes, microglia and oligodendrocytes
[33][34][35][36].
Furthermore, the brain is also temporally complex. For example, the development of the nervous system, commonly termed corticogenesis, is a highly complex process that requires the balancing of many components, including chromatin spatial organization. Disentangling the interplay of these contributing components is critical to the understanding of various diseases associated with dysfunctional cortical development, as demonstrated in Song et al.
[35]. Several other studies have also interrogated the relevance of varying developmental stages, primarily the fetal and adult brain
[34][37][38][39], to shed insights into the temporal dynamics underlying genetic regulation, and ultimate disease and health-related outcomes.
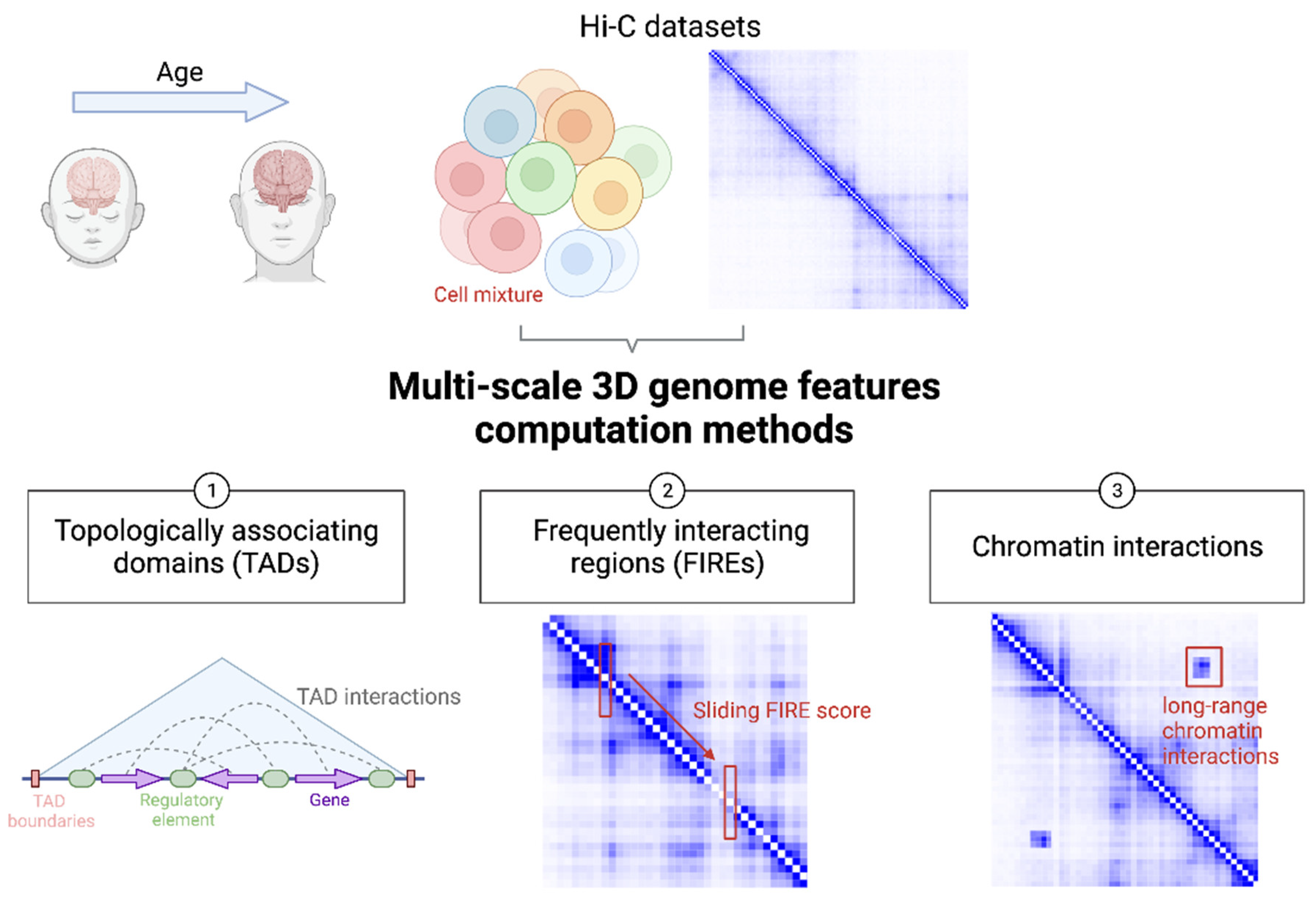
Figure 1 illustrates state-of-the-art strategies to harness the power of multi-scale readouts from brain 3D genome organization data for the understanding of genetic mechanisms underlying neuropsychiatric diseases and other brain-related traits.
Figure 1. Cartoon illustration of utilizing brain 3D genome organization data at multiple scales to understand genetic mechanisms of brain function and disease. Hi-C and like datasets can be generated from brain samples from donors at different ages. Typically, each sample contains a mixture of different brain cell types. From a typical Hi-C dataset, TADs, FIREs, and chromatin interactions can be defined at different developmental stages and from distinct cell types for comparative analysis.
3. TAD
TADs have been largely under-appreciated and under-utilized in the interpretation of genetic findings for brain-related disorders and traits, mainly because of two reasons. First, TADs, as a stable structural feature, are rather conserved across tissues, cell lines, and even across species
[13]. Second, rare structural variants (SVs) that are more likely to result in abnormal TAD formation are usually not available for analysis because prevailing genotyping arrays and short read sequencing technologies have limited capabilities to generate reliable genotypes for SVs.
Here, SCZ as an example. As a heritable disease, SCZ has been extensively studied via genotyping-array-based GWAS
[40][41][42], as well as via whole exome sequencing (WES)
[43] and whole genome sequencing (WGS)
[44]. However, WES only analyzes the protein-coding portion of the genome (around 3%), and therefore misses most regulatory regions. Previous findings
[45][46] indicate rarer and evolutionarily younger SNPs tend to have higher SNP heritability for numerous complex traits. The interrogation of the rarer regulatory variants entails WGS-based studies, which allow nucleotide-level resolution profiling of the entire genome including the vast non-coding regions. In addition, WGS empowers the detection of SVs throughout the accessible genome.
Halvorsen et al. examined the role of genetic variations that can be discovered by WGS but not by standard genotyping array or WES in the SCZ etiology
[44]. A higher genome-wide load of rare SVs including deletions (DEL), tandem duplication (DUP), inversion (INV), and mobile element insertion (MEI) sites has been identified in SCZ cases than in controls. Burden analyses of ultra-rare SVs further revealed that ultra-rare DELs are highly enriched in SCZ cases, while ultra-rare DUP and INV are neither significantly enriched nor depleted.
TAD boundary disruption by SVs has been linked to several developmental disorders
[47][48]. To help elucidate the molecular mechanisms of the enrichment of ultra-rare DELs among SCZ cases, Halvorsen et al. partitioned the elevated genome-wide burden of ultra-rare SVs among various functional elements including TADs, FIREs, ATAC-seq peaks, CTCF ChIP-seq peaks, H3K27ac ChIP-seq peaks, and H3K4me3 ChIP-seq peaks. They found that only TAD boundaries are significantly enriched with these ultra-rare SVs (
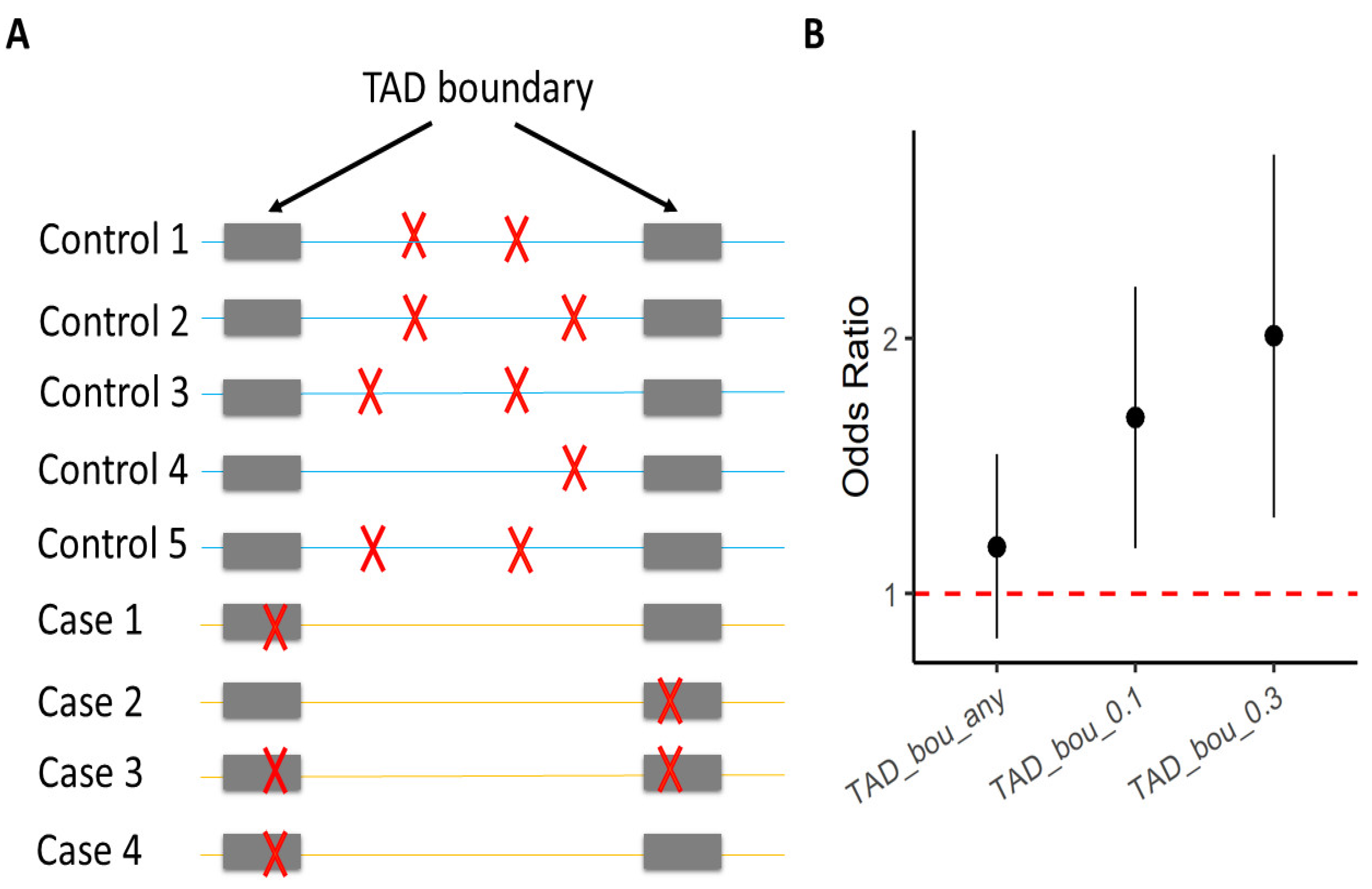
Figure 2). Specifically, their results suggest that ultra-rare SVs in SCZ cases disrupt TAD boundaries, detected from both the adult brain and the fetal brain. Modifying TAD boundaries can substantially influence enhancer–promoter interactions, and disrupt local gene expression
[47][48].
Figure 2. Enrichment of ultra-rare SVs in SCZ cases that impact TAD boundaries. (
A) Each row represents one individual; gray bars indicate TAD boundaries; red crosses mark ultra-rare SVs. (
B) The Y-axis is the odds ratio that measures the increase in the likelihood of being a SCZ case per unit increase in burden of ultra-rare SVs
[44]. The X-axis specifies different sets of ultra-rare SVs on which the burden analyses were performed. “TAD_bou_any”: ultra-rare SVs that have (≥1 bp) overlap with TAD boundaries identified from the adult brain; “TAD_bou_0.n”: ultra-rare SVs that overlap > n × 10% of TAD boundaries identified from the adult brain.
Overall, the Halvorsen et al. study provides an exemplary case where TADs are systematically assessed for their SCZ relevance. This example highlights the importance of TADs in the understanding of genetic variation linked with brain-related diseases.
4. FIRE
By analyzing a compendium of Hi-C data generated across 21 human cell lines and primary tissues, Schmitt et al.
[11] discovered FIREs as local interaction hotspots enriched for active enhancers. Crowley et al.
[38] further implemented the Poisson regression-based approach in a stand-alone computationally efficient R package FIREcaller to identify FIREs.
FIREs are chromatin features distinct from other 3D genome features such as A/B compartments, TADs, and chromatin loops, in terms of high cell type specificity. Schmitt et al., among the 21 tissues mentioned before, found that roughly 38.8% of FIREs were found in only one tissue or cell type, and about 57.7% were found in two or fewer, indicating that FIREs are highly tissue specific
[11]. FIREs are enriched in compartment A and depleted in compartment B. In addition, FIREs are depleted near TAD boundaries but are enriched within TAD and towards TAD centers. Although FIREs are enriched for chromatin loop anchors, 90% of FIREs are within chromatin loops. The dynamics of FIREs across brain developmental stages and cell types are closely associated with gene regulation dynamics during brain development and in different cell types
[36][38] (as illustrated in
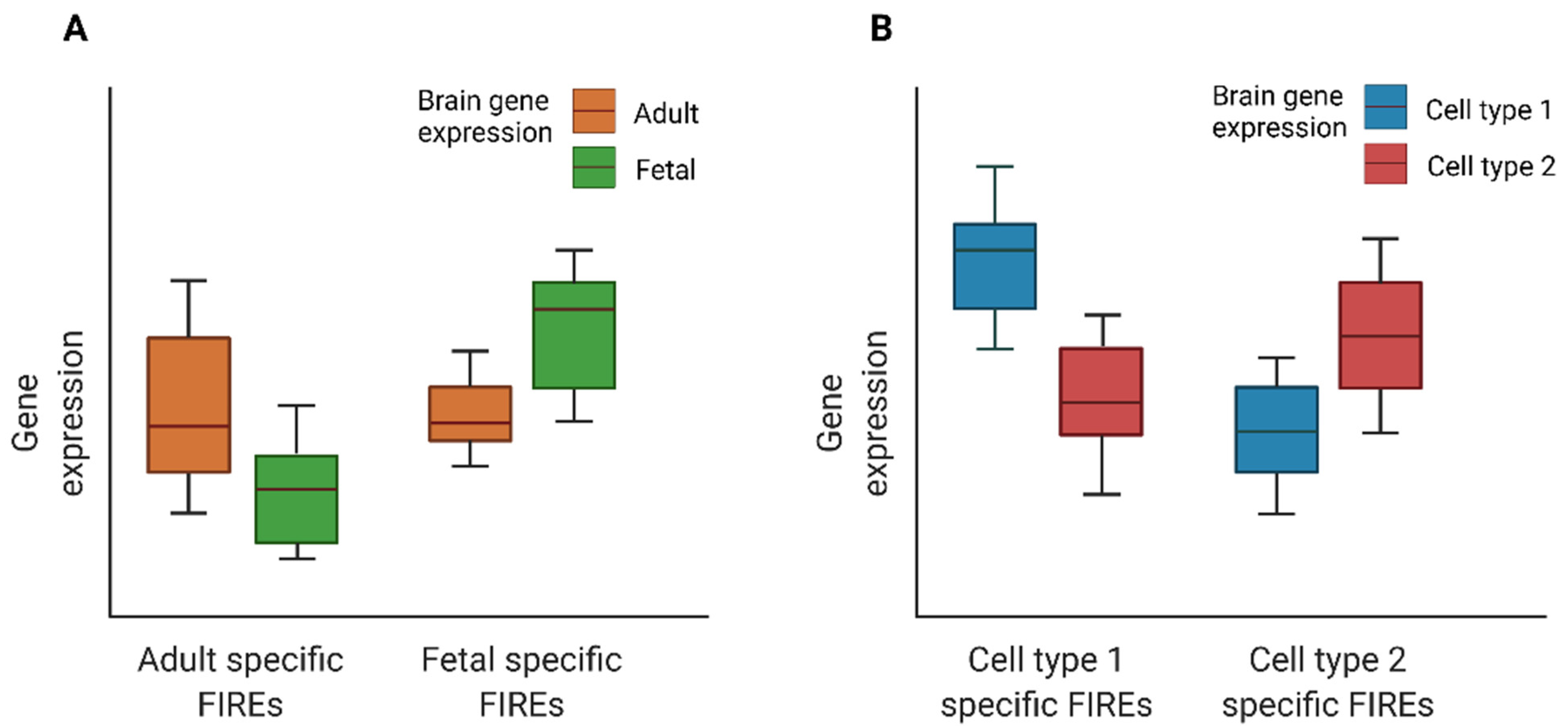
Figure 3).
Figure 3. Cartoon illustration of expression for genes overlapping developmental time- or cell type-specific FIREs: (A) Boxplots of expression for genes overlapping fetal or adult brain FIREs. (B) Boxplots of expression for genes overlapping cell type specific FIREs.
Disease candidate genes can be prioritized by examining genes near FIREs containing disease-associated GWAS SNPs
[11]. Then, by investigating the overlap between FIREs and disease-associated SNPs, Schmitt et al. found that FIREs are enriched for disease-associated GWAS SNPs
[11]. For example, they reported that an AD-associated SNP rs3851179 resides in a hippocampus-specific FIRE. Examining genes nearby, they found a putative causal gene for AD: gene
PICALM, whose 5′ end overlaps this brain-specific FIRE and is 88.5 kb away from this SNP. Another example, as shown in Crowley et al.
[38], is a SCZ-associated GWAS SNP rs9960767 residing in a hippocampus super-FIRE, which overlaps with two hippocampus super-enhancers. The gene
TCF4, to which this SNP rs9960767 is intronic, is a potential causal gene for SCZ. The hippocampus super-FIRE region within its gene body also helps to suggest the underlying regulatory mechanism.
FIREs are genomic regions that are involved in gene regulation. In a recent study, Hu et al. compared FIREs in sorted NeuN+ (representing neurons) and NeuN− (representing glia) cells to identify differential FIREs
[36]. Specifically, they defined NeuN+ specific FIREs as regions with higher FIRE scores in NeuN+ cells but lower FIRE scores in NeuN− cells; and NeuN− specific FIREs are conversely defined. Their results suggest that most NeuN+ or NeuN− specific FIREs overlap with the corresponding NeuN+ or NeuN− specific H3K27ac ChIP-seq peaks. Furthermore, genes associated with NeuN+ and NeuN− specific FIREs are primarily enriched in neurons and glial cells, respectively. These findings revealed that differentiated FIREs in the central nervous system are closely linked to cell-type-specific gene regulation. Furthermore, NeuN+ hypoacetylated and NeuN− hyperacetylated genes are enriched in co-expression modules that were downregulated and upregulated in AD, respectively. Taken together, these results suggest that FIRE-associated cell-type-specific gene regulatory networks can aid in the understanding of AD etiology.
5. Chromatin Interactions
The disruption of regulatory chromatin loops plays an important role in the etiology of complex brain disorders. For example, several studies
[49][35][50][51][52][53][54][55] have reported non-coding variants identified from genome-wide association studies (GWAS) overlapping with cis-regulatory elements that regulate distal genes by long-range chromatin interactions. It remains challenging to identify causal variants and their putative target genes in the disease relevant cell types due to the complexity of the brain tissue and the etiology underlying brain-related disorders.
Assigning variants to genes based 1D proximity provides a limited, if not sometimes misleading, view of the complexity of GWAS findings. Integrating chromatin interactome data with GWAS results can aid in the identification of potential causal variants, their effector genes, and the functional roles at each GWAS locus. For example, a recent study on AD utilized Hi-C data from fetal and adult brains to examine possible mechanisms that contribute to the regulatory effects of risk haplotypes at the
APOE locus (encompassing multiple genes including
PVRL2, APOE, and
APOC1) on the expression of nearby genes in brain tissues
[56]. They identified multiple highly interacting regions covering the risk haplotypes, suggesting broad modulatory effects of those non-coding haplotypes beyond the widely known
APOE gene at the locus. In addition, a recent SCZ study leveraged Hi-C data from the developing brain to aid in the identification of putative causal SNPs by mapping SNPs to regions identified as likely regulatory elements
[41]. Moreover, Giusti-Rodriguez et al. mapped GWAS loci associated with ten psychiatric disorders and cognitive traits, including SCZ, intelligence, attention deficit hyperactivity disorder (ADHD), alcohol dependence, AD, anorexia nervosa, autism spectrum disorder, bipolar disorder (BD), major depression disorder, and educational attainment, to thousands of genes by leveraging Hi-C data from adult and fetal brain cortex samples with concomitant RNA-seq, open chromatin (ATAC-seq), and ChIP-seq data (H3K27ac, H3K4me3, and CTCF). Linking GWAS variants to their potential effector genes helps the interpretation of identified genetic associations for these complex brain-related diseases and traits in non-coding regions
[39].
Disease-associated variants are often found in cell-type-specific enhancers, which form regulatory interactions with the promoter regions of their target genes
[43]. With the development of single cell technologies, candidate target genes can be assigned to non-coding GWAS SNPs in a cell-type-specific manner. For example, Yu et al.
[49] recently developed SnapHiC, a computational method to identify chromatin interactions from single cell Hi-C data, and leveraged cell-type-specific chromatin interactions to find putative target genes, which are likely regulated by GWAS variants associated with neuropsychiatric disorders in disease relevant cell types.
Besides Hi-C, technologies such as promoter capture Hi-C and PLAC-seq can also help elucidate genes for neuropsychiatric disorders. Song et al. found that GWAS SNPs are enriched at promoter interacting regions (PIR) in a disease- and cell-type-specific manner
[33]. Specifically, the study generated promoter capture Hi-C data for primary astrocytes and three neuronal cell types derived from induced pluripotent stem cells, from which chromatin interactions were identified in a cell-type-specific manner. They then leveraged these cell-type-specific chromatin interactions to annotate genetic variants associated with eleven complex neuropsychiatric disorders. Results showed that ASD, mental process (MP), and SCZ SNPs are enriched at PIRs across all cell types. Unipolar depression (UD) SNPs are enriched exclusively in excitatory and hippocampal dentate gyrus (DG)-like neurons, whereas AD, ADHD, and BD SNPs also exhibit enrichment in lower motor neurons. The regulatory roles of PIRs were further validated by CRISPRi experiments
[33].
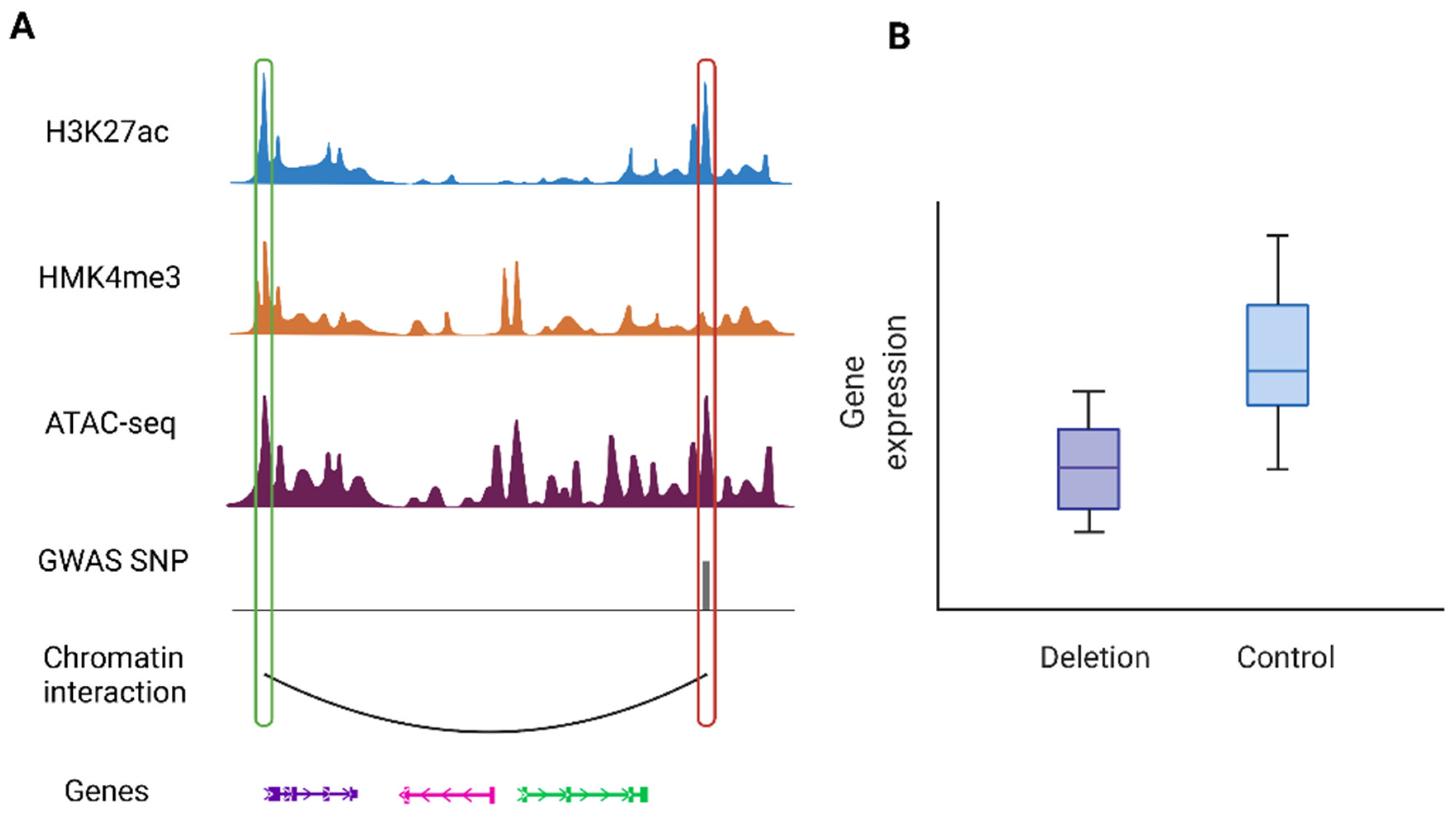
Figure 4 shows an illustration of such cases where a PIR containing a disease risk variant interacts with the promoter of its target gene to regulates gene expression. Likewise, analysis of human post-mortem cortical tissue shows that sporadic AD variants are largely involved in microglia-specific chromatin interactions, while variants associated with various neuropsychiatric disorders are primarily confined to neuronal-specific enhancer-promoter networks
[34].
Figure 4. An illustrative example of a chromatin interaction involving disease-associated variants: (A) An example of a PIR containing a risk variant (highlighted by the red box on the right) with promoter region of the target gene (highlighted by the green box on the left). (B) Regulatory roles of the risk variant can be validated by downstream experiments such as CRISPR techniques. This example shows deletion of the PIR containing the risk variant results in downregulation of the target gene (the left most gene).
Few studies have investigated the impact of larger-scale or more complex rearrangements of DNA sequences on chromatin interactions because these rearrangements are much less well characterized than SNPs
[57][58]. For example, Johnston et al.
[59] observed many chromatin interactions involving DNA segments >1 Mb apart or even >100 Mb, in their Hi-C data from glioblastoma stem cells. Hypothesizing that SVs may explain these surprisingly long-range interactions, they explicitly studied interactions involving SVs or not, finding that the apparent distance of interactions involving SVs is >10×that of interactions without SVs in the neighborhood. They presented a representative example at the
JAK1 locus where a 140 Mb inversion together with other large deletions moved two enhancers residing normally on the q-arm of chromosome 1 to the p-arm near the
JAK1 gene, leading to chromatin interactions that would be normally impossible. Similarly, Wang et al.
[60] reported enhancer hijacking that resulted from SVs in their Hi-C data from cell lines derived from patients affected with pediatric high-grade gliomas.