Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gaspare La Rocca | -- | 3991 | 2022-04-06 16:37:22 | | | |
| 2 | Jessie Wu | -21 word(s) | 3970 | 2022-04-07 03:23:03 | | | | |
| 3 | Jessie Wu | Meta information modification | 3970 | 2022-04-07 03:43:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
La Rocca, G.; Cavalieri, V. Roles of the miRISC's Components in Chromatin Biology. Encyclopedia. Available online: https://encyclopedia.pub/entry/21423 (accessed on 27 July 2026).
La Rocca G, Cavalieri V. Roles of the miRISC's Components in Chromatin Biology. Encyclopedia. Available at: https://encyclopedia.pub/entry/21423. Accessed July 27, 2026.
La Rocca, Gaspare, Vincenzo Cavalieri. "Roles of the miRISC's Components in Chromatin Biology" Encyclopedia, https://encyclopedia.pub/entry/21423 (accessed July 27, 2026).
La Rocca, G., & Cavalieri, V. (2022, April 06). Roles of the miRISC's Components in Chromatin Biology. In Encyclopedia. https://encyclopedia.pub/entry/21423
La Rocca, Gaspare and Vincenzo Cavalieri. "Roles of the miRISC's Components in Chromatin Biology." Encyclopedia. Web. 06 April, 2022.
Copy Citation
The Argonaute (AGO) and the Trinucleotide Repeat Containing 6 (TNRC6) family proteins are the core components of the mammalian microRNA-induced silencing complex (miRISC), the machinery that mediates microRNA function in the cytoplasm. The cytoplasmic miRISC-mediated post-transcriptional gene repression has been established as the canonical mechanism through which AGO and TNRC6 proteins operate. However, growing evidence points towards an additional mechanism through which AGO and TNRC6 regulate gene expression in the nucleus.
miRISC
Argonaute
piRNA
1. Evolutionary Conservation of AGO-Mediated TGS Mechanisms
The isolation of the RITS in Schizosaccharomyces pombe S. pombe). From yeast to humans, heterochromatin constitutes a group of transcriptionally silent chromatin domains essential for the maintenance of chromosome integrity and for the regulation of gene expression [1]. Heterochromatin regions are marked by the presence of members of the heterochromatin protein 1 (HP1) family that bind to repressive histone marks, such as di- and trimethylated histone H3 lysine 9 (H3K9me2 and H3K9me3, respectively) [2] through a conserved N-terminal domain named the chromodomain. H3K9 methylation spreads along chromatin through sequential cycles of methylation, mediated by H3K9 methyltransferase enzymes, coupled to the oligomerization of HP1 family members [3]. A link between AGO proteins and heterochromatin remodeling has been extensively characterized in S. pombe. The mechanistic basis of this link began to be elucidated with the isolation of the RNA-induced transcriptional silencing (RITS) nuclear complex, whose core components are Chp1 (a chromodomain-containing protein), Ago1, a Dicer-dependent sRNA, and Tas3 [4], a protein that contains a GW-rich domain with “AGO-hook” properties [5]. The RITS was shown to mediate heterochromatin formation at centromeric and pericentromeric DNA sequences complementary to the Ago1-associated sRNAs through TGS (Figure 1A). Interestingly, the sRNAs loaded on the RITS, often referred to as repeat-associated siRNAs (rasiRNAs) [6], are produced by the Dicer-mediated processing of long dsRNAs that results from convergent antisense transcription of the same centromeric regions where heterochromatin assembly initiated [7]. Deletion of any of the RITS component genes, or of dicer with the consequent loss of rasiRNAs, induces defects in heterochromatin maintenance. Once bound to the chromatin, the RITS recruits the RNA-dependent RNA polymerase complex (RDRC) formed by Rdp1 polymerase, the helicase Hrr1, and the non-canonical poly(A) polymerase Cid12. The RDRC complex amplifies the silencing process initiated by RITS in a positive feedback loop consisting of the further synthesis of target-specific dsRNA, their processing by Dicer and subsequent production of rasiRNA, and additional recruitment of more RITS to the region [8] (Figure 1A).
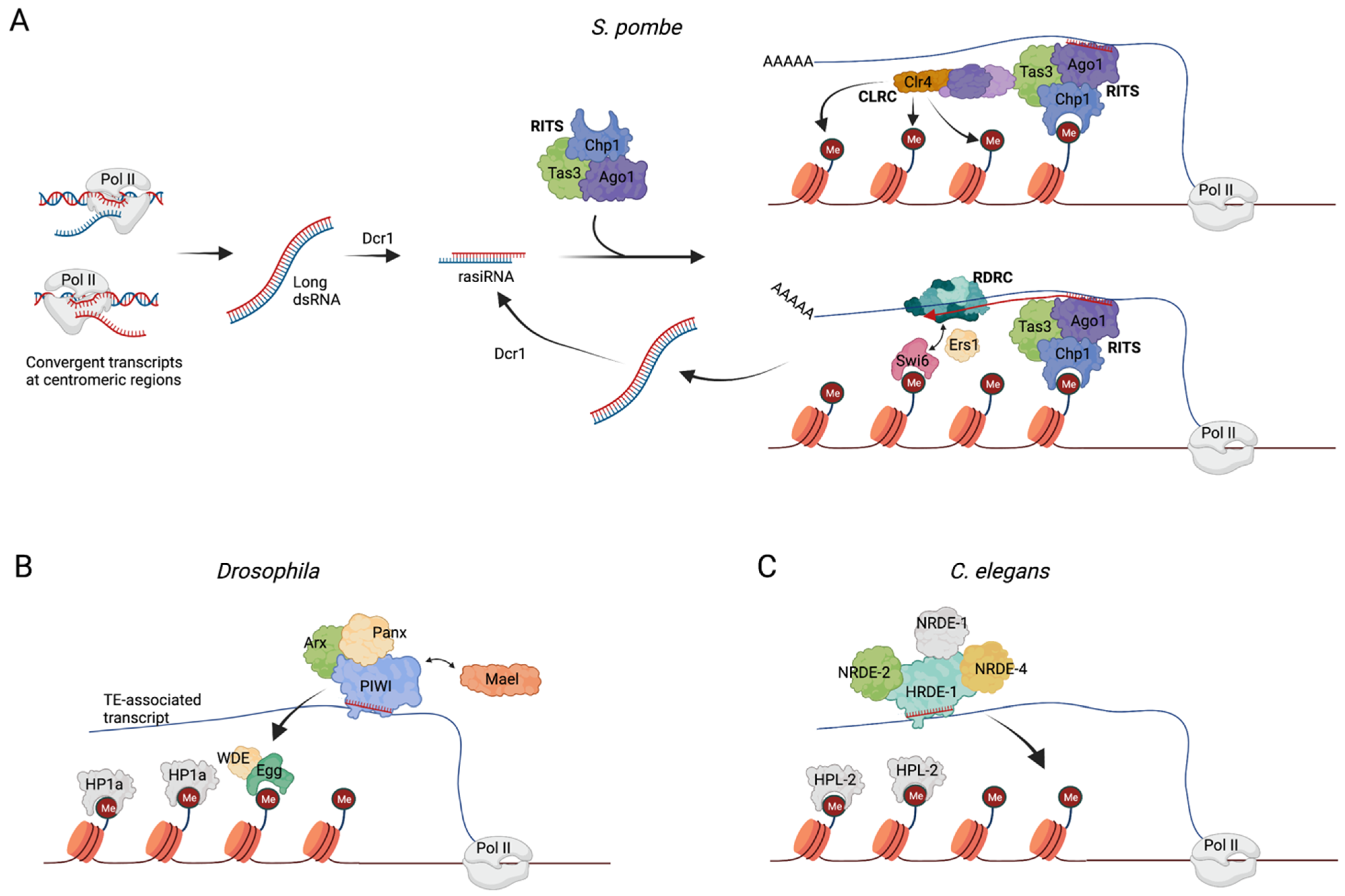
Figure 1. (A) The RITS complex in S. pombe. Convergent transcription at the targeted centromeric regions generates long double strand RNAs which are cleaved by Dicer1 (Dcr1) into small 25–30 nt dsRNAs, named repeat-associated siRNAs (rasiRNAs). One strand of the rasiRNA is loaded onto the RITS, and the resulting ribonucleoprotein complex is recruited on nascent transcripts spanning the targeted centromeric region. On the nascent transcript, the RITS can associate both with the CLRC complex to induce histone methylation, and with the RDRC complex to produce additional targeting rasiRNAs. RDRC recruitment is facilitated by Ers1, which acts as a bridge between the RDRC subunit Hrr1 and the chromodomain protein Swi6 [9]. (B) In Drosophila, Piwi–piRNA complexes bind TE-associated nascent transcripts, a process in which the proteins Arx and Panx take part. The histone methyltransferase, Egg, and the accessory protein, Wde, deposit H3K9me2/3 marks on transposon sequences, a process assisted by Mael. Next, HP1a binds to the targeted locus to maintain gene repression. (C) Mechanism of TGS in C. elegans operated by HRDE-1/WAGO-9 protein (details described in the main text).
Although the RITS can, in principle, recognize DNA sequences to target specific chromatin regions, several studies have supported a model according to which the sequences within a transcript generated from the targeted locus act instead as the docking site for the rasiRNA-loaded Ago1 [8][10][11][12] (Figure 1A).
In addition to the nascent transcript and the RDRC and RITS complexes, the heterochromatin assembly platform is also comprised of the CLRC complex, which is required for H3K9-specific methylation and heterochromatin maintenance. The core components of CLRC are the histone methyltransferase (HMT) Clr4, the Cullin scaffold protein Cul4, the RING finger protein Pip1, the DNA damage binding protein 1 homolog Rik1, and the DCAF-like protein delocalization of Swi6 1 and 2 (Dos1 and Dos2) [13][14][15][16][17]. A LIM domain protein, Stc1, mediates the interaction between CLRC and the RITS complexes, and is required for RNA-dependent H3K9 methylation [18]. The RITS, RDRC, and CLRC complexes physically interact, and conceivably work in synergy to establish and maintain heterochromatin structure.
The mechanism underlying Ago-mediated heterochromatic modifications in plants resembles the mechanism in S. pombe. For example, in Arabidopsis thaliana, the DICER-LIKE proteins DCL1 to 4 produce sRNAs that are loaded into 1 of the 10 Argonaute proteins encoded by the genome of this organism. Of note, in plants, sRNA-induced TGS relies principally on DNA methylation at cytosine residues by the RNA-directed DNA methylation pathway (RdDM). RdDM directs de novo DNA methylation while other DNA methyltransferases, such as MET1 or CMT3, mediate DNA methylation maintenance [19].
The piRNA pathway. Mechanisms of TGS are also present in animals. TGS in animals is best understood in the piRNA pathway, where the Piwi clade of Argonautes direct the silencing of transposable elements (TE) in gonadal tissues [20][21]. In Drosophila, Piwi proteins/piRNA complexes operate in the nucleus where they recognize nascent transposon transcripts and direct the TGS of the corresponding transposon loci [22][23]. Recognition of the nascent transcript by Piwi–piRNA complexes is stabilized by the Piwi-interacting zinc-finger protein Asterix (Arx) [24]. Panoramix (Panx), a protein with no known domain [24][25][26], is also a key TGS effector, which acts at the interface between the piRNA pathway and the chromatin silencing machinery [23][26]. Downstream of Arx and Panx, the coordinated action of dLsd1/Su(var)3–3 and Eggless/dSETDB1 leads to the removal of H3K4me2 and to the establishment of H3K9me3, respectively, followed by heterochromatin formation via the recruitment of HP1a/Su(var)205 [21][27][28][29] (Figure 1B).
Similar mechanisms govern TGS in C. elegans. In this organism, sRNAs named 22G-RNAs repress transposable elements in the germline [30][31]. The Argonaute protein HRDE-1/WAGO-9, loaded with 22G-RNAs, shuttles to the nucleus and recognizes nascent RNA transcripts. The coordinated action of additional factors, such as NRDE-1, -2, and -4, results in RNA pol II stalling, H3K9me3 deposition, and the binding of HPL-2—a HP1a homolog—ultimately resulting in the transcriptional silencing of the target locus [32][33] (Figure 1C).
Although the piRNA pathway has been primarily described as a mechanism to repress transposons, a large portion of identified piRNAs in C. elegans does not overlap with known transposon sequences [34], suggesting other possible regulatory functions mediated by the piRNA pathway. Growing evidence shows that piRNAs are indeed involved in the global regulation of endogenous transcriptional programs. For example, Cornes et al. have recently reported that piRNAs mediate the transcriptional silencing of endogenous genes to promote sperm differentiation [35].
As described above, TGS mechanisms in evolutionarily distant species share common mechanisms relying on: (1) the recognition of nascent transcripts by an Argonaute family loaded with sRNA; (2) the recruitment of heterochromatin factors; and (3) silencing of the targeted loci. As described in the next section, similar mechanisms also take place in mammals.
2. Evidence of TGS in Mammals
TGS triggered by exogenous sRNAs. Several examples of TGS in mammals have been described (Table 1). Early studies in cell lines have shown that sRNAs that are exogenously delivered could inhibit gene expression and modify chromatin [36][37][38][39]. For example, synthetic sRNAs that are complementary to sequences within the elongation factor 1 alpha (EF1A) promoter could repress the expression of an integrated EF1A promoter–reporter transgene, and, although to a lesser extent, the expression of the endogenous EF1A locus [37]. In this model, gene silencing was associated with cytosine DNA methylation (henceforth DNA methylation, a hallmark of silent chromatin), which was abolished by treatment with 5-azacytidine, an inhibitor of DNA methyltransferases, indicating that gene silencing occurred entirely through a mechanisms similar to the TGS described in S. pombe, and did not involve RNA-mediated PTGR events [37].
However, it became clear that DNA methylation was not always associated with sRNA-mediated TGS [39][40][41]. For example, the absence of DNA methylation during TGS triggered by exogenous sRNAs was also observed in the case of the progesterone receptor (PR) gene [41]. Ting at al. have shown that, although sRNAs directed to the E-cadherin (CDH1) promoter could promptly induce transcriptional repression, this process could take place even in cells depleted of virtually any capacity to perform DNA methylation [39].
While the extent to which DNA methylation participates in sRNA-induced TGS is still under debate, more compelling evidence has been provided for sRNA-induced silencing through repressive histone modifications [37][42]. For example, Weinberg et al. showed a fourfold increase in H3K9 and H3K27 methylation in proximity of the endogenous EF1A promoter upon transfection of promoter-directed sRNAs. Interestingly, H3K9 methylation was even induced at sequences hundreds of bps downstream of the EF1A promoter, suggesting that the sRNA-mediated targeting may induce the spreading of chromatin marks to regulate neighboring regions [42].
The observation that promoter-directed sRNAs could induce chromatin changes in mammalian cells ignited interest towards the factors that could mediate this process. The structural and functional information previously gathered from the RITS and the piRNA pathways in Drosophila and C. elegans inspired the search for a potential role of AGO as the physical interface between sRNAs and the chromatin-modifying machinery. Such a role for AGO came to light in chromatin immunoprecipitation (ChIP) experiments in human cells. These studies showed that the intracellular delivery of sRNA could induce up to a 20-fold increase of AGO1 and H3K9me2 presence in proximity of the sequences complementary to the sRNA [43]. Interestingly, H3K9me2 was detectable up to 300 bp downstream of the sRNA-binding site, suggesting that the AGO–sRNA complex, by recruiting histone methyltransferases (HMTs), initiates the formation of heterochromatic foci, which eventually propagate to the surrounding regions. AGO1-directed TGS and H3K9me2 formation required protein–protein interactions between AGO1 and both TRBP2 and Pol II, and was accompanied by an enrichment of the HMT EZH2, a Polycomb component responsible for the synthesis of H3K27me3. Within this multi-subunit complex, the role of the TRBP2 is not fully understood, although it may mediate the loading of the guide sRNA on AGO, similar to its role in the miRLC in the cytoplasm [44]. The recruitment of EZH2 onto the sRNA-targeted site may be necessary for H3K27me3 accumulation, linking AGOs to the endogenous mechanisms of Polycomb silencing. Finally, the requirement of Pol II is consistent with the formation of a nascent transcript that acts as the landing platform for the AGO1–sRNA complex, although the role of such a transcript was not explored in detail in this early study.
Recent studies have further elucidated the identity and modus operandi of the enzymatic activities recruited by AGO during TGS, although the high complexity of mammalian genomes has made the search for these enzymes more challenging than the search previously undertaken in S. pombe. For example, while Clr4 is the only H3K9-specific methyltransferase within the RITS in S. pombe, mammalian genomes encode for several HMTs with H3K9 specificity, any of which could, in principle, function during RNA-induced TGS. So far, among these enzymes a role in TGS has only been documented for SETDB1, as evidenced in a study in which sRNAs directed to the androgen receptor (AR) promoter increased the local recruitment of both AGO2 and SETDB1 around the sRNA binding site [45]. Moreover, EZH2 was also enriched on the targeted promoter and was physically associated with SETDB1, suggesting that the two complexes may cooperate during TGS in mammalian cells. SIN3A and HDAC2, key members of the SIN3–HDAC corepressor complex, whose function is to promote histone deacetylation and heterochromatin formation, are also enriched at the targeted promoter in a AGO2- and SETDB1-dependent manner and physically interact with SETDB1 [45]. These observations are consistent with a model where the SIN3–HDAC corepressor complex cooperates with AGO2 and SETDB1 in the establishment of silent chromatin in regions surrounding the sRNA target site. Importantly, the recruitment of SETDB1–AGO2 and the formation of repressive histone marks within the region surrounding the AR promoter required a nascent ncRNA spanning the promoter region. This observation provides further evidence that a nascent RNA transcript may be the preferential recognition platform for sRNA-guided chromatin remodeling of the template DNA. Notably, DNA methylation is not induced following targeting of the AR promoter, consistent with the notion that the induction of repressive histone modifications may play a greater role in mammalian TGS.
miR-dependent pathways of TGS. The discovery that exogenous sRNAs could induce TGS and chromatin silencing in mammalian cells led to the speculation that endogenous sRNAs with analogous function may exist. miRs were an obvious class of candidates to explore, given their similarity in size with the RITS-associated sRNAs. Early attempts to discover an example of miRs-mediated TGS relied on computational tools that search for sequences within gene promoters with extensive complementarity to endogenous miRs. This approach led to numerous examples of miR-induced TGS in mammals. Authors of the first report of miR-mediated TGS in mammalian cells showed that miR-320 is generated within the promoter of the cell cycle gene POLR3D, and its depletion induced an increase of POLR3D expression at the transcriptional level [46]. Moreover, the overexpression of miR-320 in HEK-293 cells induced an enrichment of AGO1, EZH2, and H3K27me3 onto the POLR3D promoter, consistent with a role of miR-320 in the induction of repressive chromatin at the targeted site. Sense transcripts spanning the POLR3D promoter were also detected, although authors have not investigated their role in AGO1 recruitment and gene repression. Given the central role of POLRD3 in the cell cycle progression [47], the potential implication of AGO-mediated TGS in proliferative disorders were highlighted.
Using computational methods to screen for miR target sites within gene promoters, Younger et al. found multiple miRs that were able to target the promoter of the human progesterone receptor (PR) and of immunoglobulin superfamily member 1 (IGSF1). Among those, the overexpression of miR-423-5p was shown to decrease Pol II occupancy, and to induce accumulation of H3K9me2 at the promoters of the two mentioned genes, in a DNA-methylation-independent manner. Moreover, TGS required the recruitment of AGO2 onto a ncRNA-encompassing gene promoter [48]. Of note, some studies using exogenous sRNAs directed to the PR promoter have ruled out a role for TNRC6 in the TGS of this locus [49].
More recently, Di Mauro et al. have found a link between Wnt signaling and miR-mediated epigenetic regulation in cardiac cells [50]. They have found that, upon drug-elicited inhibition of the Wnt pathway, the level of the cardiac-enriched miR-133a increases in the nucleus of HL-1 cardiac cells. Importantly, a knock-down of AGO2 (but not AGO1) and IMPORTIN-8 abrogated re-localization, suggesting a role of these two proteins in the shuttling of miR-133a into the nucleus. Enrichment of miR-133a in the nucleus coincided with the downregulation of several transcription factors and epigenetic enzymes, including DNMT3b, whose promoter contains a binding site for miR-133a. These authors were able to directly link the nuclear enrichment of miR-133a with the transcriptional silencing of DNMT3, as the transfection of cells with antisense oligonucleotides against miR-133a binding sites prevented DNMT3b repression. ChIP analysis showed that the inhibition of Wnt signaling was also accompanied by AGO2 localization at the wnt promoter, an increase in the histone repressive mark H3K27me3, and a decrease of H3K4me3 that marks active transcription. Interestingly, DNMT3B was also recruited at the miR-133a binding site and was physically associated with AGO2. As a consequence of the recruitment of DNMT3B, DNA methylation levels at the promoter increased, forming a negative feedback mechanism through which the miR-mediated recruitment of DNMT3B resulted in the repression of its own expression.
Endogenous miRs have also been involved in the transcriptional repression of the metastasis-promoting gene matrix metalloproteinase 14 (MMP-14). This gene is activated by the transcription factor Yin Yang 1 (YY1), and its overexpression or downregulation promotes or inhibits tumor progression, respectively. Zheng et al. noted that a YY1 binding site is located just 30–40 bp downstream of a putative binding site for miR-584-3p within the MMP-14 promoter, suggesting a possible functional interaction between the two elements. Indeed, in gastric cancer cells, the authors found that miR-584-3p binds to the MMP-14 promoter in an AGO2-dependent manner, leading to increased levels of the repressive histone marks H3K27me3 and H3K9me2. The induction of heterochromatin hinders the recruitment of YY1 and consequently suppresses MMP-14 expression. Importantly, treatment with RNase H reversed miR-584-3p-mediated chromatin modification, indicating that miR-584-3p directly interacted with the MMP-14 promoter via an RNA–DNA hybrid. By impeding the binding of YY1 to the MMP-14 promoter, an AGO2–miR-584-3p complex can suppress the tumorigenesis and aggressiveness of gastric cancer cells, both in cell lines and xenograft tumors [51].
It is unclear how widespread miR-mediated TGS occurs within mammalian gene networks, and to what extent either DNA or RNA acts as a recruitment platform. Nevertheless, the expression of ncRNA-spanning promoter regions seems to be a common feature of several mammalian promoters [52][53], which makes them potential targets of miR-mediated TGS mechanisms. The repurposing of target prediction algorithms to scan such promoter sequences for sites complementary to miRs is certainly a valid approach to potentially predict new examples of miR-mediated TGS [54][55].
3. miR-Dependent Endogenous Pathways of TGA in Mammals
TGA mechanisms act through a promoter and enhancer. The first evidence that endogenous miRs can induce TGA came from a study in which the transcription of the CDH1 and CSDC2 genes, whose promoters contain sequences complementary to miR-373, was upregulated by the overexpression of miR-373. This early study, although it provided evidence for endogenous pathways that mediate TGA, did not investigate the potential involvement of AGO proteins or nascent transcripts in this process [56].
The mechanistic details on miR-mediated TGA were soon provided by Huang at al. By using a combination of in silico and wet lab approaches, these authors found that miR-744 and miR-1186 can bind to complementary sites within the CCNB1 promoter and subsequently enhance CCNB1 expression in mouse cell lines. ChIP analysis revealed that the expression of miR-744 and miR-1186 induced the localization of AGO1 on the CCNB1 promoter, accompanied by increased levels of RNA Pol II and H3K4me3 at the CCNB1 TSS [57]. Since CCNB1 overexpression promotes tumorigenesis [58][59], and the overexpression of miRs miR-744 and miR-1186 phenocopies CCNB1 overexpression, this work indicates that TGA may be a process through which miRs can contribute to cancer development.
Further studies have similarly supported a role of TGA in tumor formation and progression. Majid et al. showed that miR-205 binds to complementary sites within the promoters of the tumor suppressor genes IL24 and IL32, enhancing their expression via TGA. This process led to induced apoptosis and cell cycle arrest, and impaired the invasive properties of prostate cancer cells. Accordingly, the overexpression of miR-205 was accompanied by an enrichment of Pol II, H3K4me2, and H3/H4ac in the promoter of both genes, indicating transcriptional activation [60]. However, although a role of AGO proteins in this instance is conceivable, this aspect was not formally addressed.
By using neuroblastoma cells as a model, Qu et al. found that the overexpression of miR-558 induced the expression of the heparanase (HPSE) gene, which was accompanied by a corresponding decrease of H3K9me2 and H3K27me3, and an increase of H3K4me3 and RNA Pol II on the HPSE promoter. HPSE gene activation requires the presence of AGO1, but not AGO2, as only a knock-down of AGO1 abolished the miR-558-induced enrichment of active epigenetic markers on the promoter.
Although RNase-H treatment reduced TGA on the HPSE promoter, suggesting that the AGO1–miR-558 complex relies on DNA–RNA pairing for promoter recognition, such a mechanism has not been thoroughly investigated and no direct evidence has been provided for this model of targeting [61].
miRs can mediate TGA by promoting a favorable chromatin status not only at the promoters, but also at the enhancers of target genes. Xiao et al. observed that about 25% of loci involved in the production of nuclear-enriched miRs overlap with markers of active enhancers, namely p300/CBP, H3K4me1, and H3K27ac, as well as Dnase I-sensitive regions. Moreover, they observed that the expression of this subgroup of miRs positively correlated with the expression of surrounding loci. These observations prompted them to test the hypothesis that miRs may target enhancers overlapping their own loci and, in turn, activate both their own expression and the expression of surrounding genes. This hypothesis was confirmed by ectopically overexpressing miR-24-1, whose locus maps within a region with enhancer function. Indeed, the overexpression of miR-24-1 led to the expression of the endogenous miR-24-1, as well as the expression of its neighbor protein-coding genes FBP1 and FANCC. TALEN deletion of the enhancer blocked the ability of miR-24-1 to activate FBP1 and FANCC. Importantly, TGA on this locus was abolished in a AGO2-null background, implying that miR-24-1-dependent TGA is mediated by AGO2. Moreover, miR-24-1 overexpression induced an enrichment of Pol II and the consequential transcription of RNAs overlapping the enhancer region, concomitant with increased levels of AGO2, p300, H3K27ac, and H3K4me1, as well as a reduced level of H3K9me3. Notably, the overexpression of miR24-1 not only led to an increased H3K27ac within the proximal enhancer, but also at thousands of enhancers with sequences complementary to the miR-24-1 [62].
It is also important to note here that, although promoters and enhancers seem to be the preferential target sites for TGA in mammals, the possibility that other genomic regions may also be recognized by AGO–miR complexes to initiate permissive chromatin has yet to be determined.
The purification of the RNA-induced transcriptional activation complex. Recently, a complex able to mediate TGA has been characterized. Using the human CDKN1A (p21) as a model gene, Portnoy et al. have isolated the RNA-induced transcriptional activation (RITA) complex, which, in addition to the saRNA–AGO2 complex, includes the nuclear DNA helicase II (RHA) and CTR9 [63]. RHA is known to bind to DNA and mediate transcriptional activation by recruiting basal transcription machinery to promoter DNA and/or modifying chromatin structure [64]. The presence of RHA in the RITS may suggest that the targeting of this promoter by saRNA involves the recognition of genomic DNA. However, this aspect is not clear, and the recognition of a nascent transcript may still be the preferential targeting mechanism in this context (Figure 2). CTR9 is a component of PAF1C, which is required to recruit histone-modifying factors such as E2/E3 ubiquitin ligase to the Pol II complex to mediate H2B ubiquitination, and, consequently, H3K4 and H3K79 methylation [65]. RITA also recruits RNA Pol II, leading to productive transcription elongation, which highlights its polyvalent role in activating gene expression. TNRC6 and components of the miRISC-loading complex (DICER, TRBP) were absent from the RITA complex, suggesting that the AGO2 pool employed in TGA associates with a unique set of proteins distinct from those found in the cytoplasmic miRISC. However, as described below, there are instances in which TNRC6 can associate with AGO proteins to mediate TGA in mammalian cells, leading to the speculation that its requirement in this process may be context- or gene-specific.
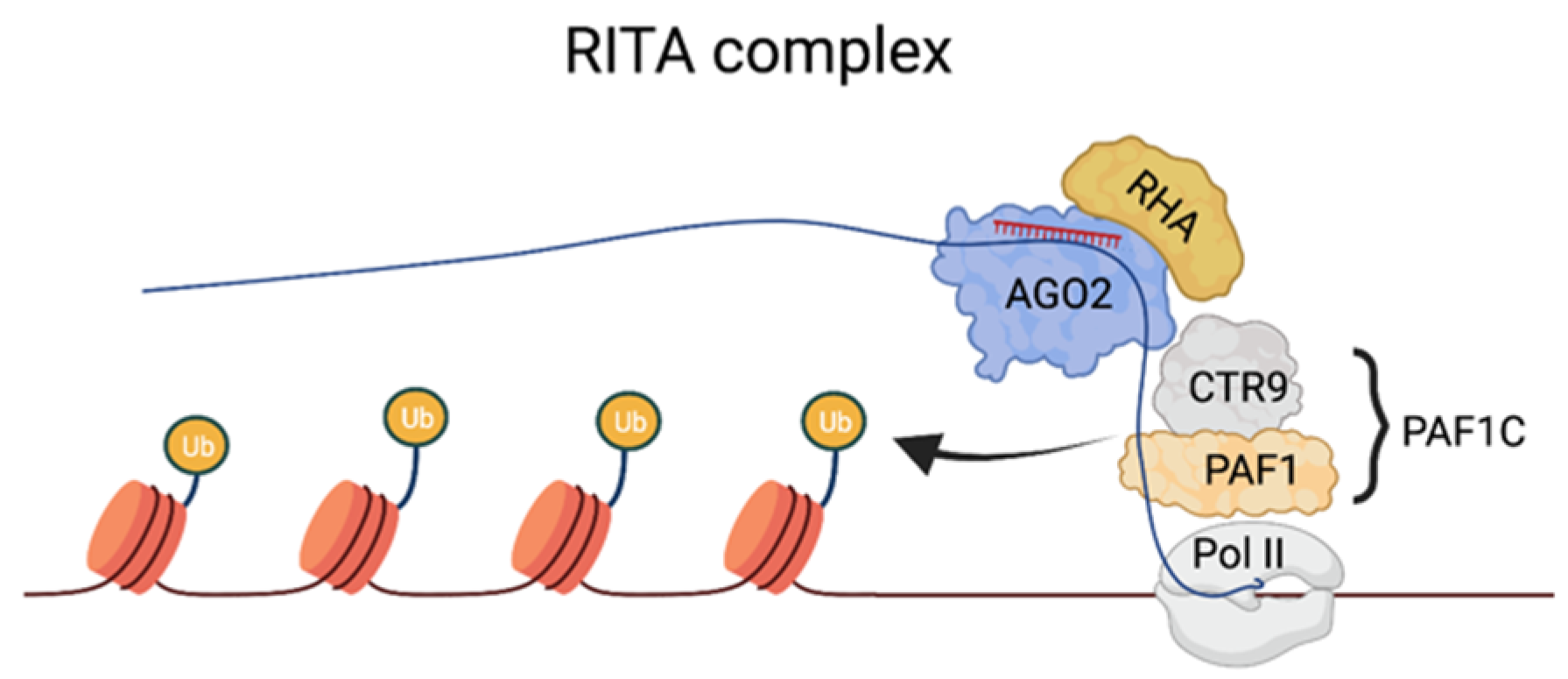
Figure 2. The RITA complex and the mechanisms of TGA at the p21 promoter. Binding sites on a nascent transcript overlapping the target are recognized by the saRNA–AGO2 complex. saRNA–AGO2 serves as a docking platform for components of PAF1C, which stimulate transcription initiation and recruit histone-modifying factors, such as E2/E3 ubiquitin ligase, to the Pol II complex to mediate H2B ubiquitination.
References
- Allshire, R.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2017, 19, 229–244.
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596.
- Grewal, S.I.S.; Moazed, D. Heterochromatin and Epigenetic Control of Gene Expression. Science 2003, 301, 798–802.
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.S.; Moazed, D. RNAi-Mediated Targeting of Heterochromatin by the RITS Complex. Science 2004, 303, 672–676.
- Till, S.; Lejeune, E.; Thermann, R.; Bortfeld, M.; Hothorn, M.; Enderle, D.; Heinrich, C.; Hentze, M.; Ladurner, A. A conserved motif in Argonaute-interacting proteins mediates functional interactions through the Argonaute PIWI domain. Nat. Struct. Mol. Biol. 2007, 14, 897–903.
- Bühler, M.; Spies, N.; Bartel, D.P.; Moazed, D. TRAMP-mediated RNA surveillance prevents spurious entry of RNAs into the Schizosaccharomyces pombe siRNA pathway. Nat. Struct. Mol. Biol. 2008, 15, 1015–1023.
- Yu, R.; Jih, G.; Iglesias, N.; Moazed, D. Determinants of Heterochromatic siRNA Biogenesis and Function. Mol. Cell 2013, 53, 262–276.
- Motamedi, M.R.; Verdel, A.; Colmenares, S.U.; Gerber, S.; Gygi, S.P.; Moazed, D. Two RNAi Complexes, RITS and RDRC, Physically Interact and Localize to Noncoding Centromeric RNAs. Cell 2004, 119, 789–802.
- Hayashi, A.; Ishida, M.; Kawaguchi, R.; Urano, T.; Murakami, Y.; Nakayama, J.-I. Heterochromatin protein 1 homologue Swi6 acts in concert with Ers1 to regulate RNAi-directed heterochromatin assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 6159–6164.
- Buehler, M.; Verdel, A.; Moazed, D. Tethering RITS to a Nascent Transcript Initiates RNAi- and Heterochromatin-Dependent Gene Silencing. Cell 2006, 125, 873–886.
- Bühler, M.; Haas, W.; Gygi, S.P.; Moazed, D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell 2007, 129, 707–721.
- Shimada, Y.; Mohn, F.; Bühler, M. The RNA-induced transcriptional silencing complex targets chromatin exclusively via interacting with nascent transcripts. Genes Dev. 2016, 30, 2571–2580.
- Hong, E.-J.E.; Villen, J.; Moazed, D. A Cullin E3 Ubiquitin Ligase Complex Associates with Rik1 and the Clr4 Histone H3-K9 Methyltransferase and is Required for RNAi-Mediated Heterochromatin Formation. RNA Biol. 2005, 2, 106–111.
- Horn, P.J.; Bastie, J.-N.; Peterson, C.L. A Rik1-associated, cullin-dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes Dev. 2005, 19, 1705–1714.
- Jia, S.; Kobayashi, R.; Grewal, S.I.S. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat. Cell Biol. 2005, 7, 1007–1013.
- Li, F.; Goto, D.B.; Zaratiegui, M.; Tang, X.; Martienssen, R.; Cande, W.Z. Two novel proteins, dos1 and dos2, interact with rik1 to regulate heterochromatic RNA interference and histone modification. Curr. Biol. 2005, 15, 1448–1457.
- Thon, G.; Hansen, K.R.; Altes, S.P.; Sidhu, D.; Singh, G.; Verhein-Hansen, J.; Bonaduce, M.J.; Klar, A.J.S. The Clr7 and Clr8 Directionality Factors and the Pcu4 Cullin Mediate Heterochromatin Formation in the Fission Yeast Schizosaccharomyces pombe. Genetics 2005, 171, 1583–1595.
- Bayne, E.H.; White, S.A.; Kagansky, A.; Bijos, D.A.; Sanchez-Pulido, L.; Hoe, K.-L.; Kim, D.-U.; Park, H.-O.; Ponting, C.P.; Rappsilber, J.; et al. Stc1: A Critical Link between RNAi and Chromatin Modification Required for Heterochromatin Integrity. Cell 2010, 140, 666–677.
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506.
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2018, 20, 89–108.
- Czech, B.; Preall, J.B.; McGinn, J.; Hannon, G.J. A Transcriptome-wide RNAi Screen in the Drosophila Ovary Reveals Factors of the Germline piRNA Pathway. Mol. Cell 2013, 50, 749–761.
- Klenov, M.S.; Sokolova, O.A.; Yakushev, E.Y.; Stolyarenko, A.D.; Mikhaleva, E.A.; Lavrov, S.A.; Gvozdev, V.A. Separation of stem cell maintenance and transposon silencing functions of Piwi protein. Proc. Natl. Acad. Sci. USA 2011, 108, 18760–18765.
- Sienski, G.; Dönertas, D.; Brennecke, J. Transcriptional Silencing of Transposons by Piwi and Maelstrom and Its Impact on Chromatin State and Gene Expression. Cell 2012, 151, 964–980.
- Donertas, D.; Sienski, G.; Brennecke, J. Drosophila Gtsf1 is an essential component of the Piwi-mediated transcriptional si-lencing complex. Genes Dev. 2013, 27, 1693–1705.
- Sienski, G.; Batki, J.; Senti, K.-A.; Dönertas, D.; Tirian, L.; Meixner, K.; Brennecke, J. Silencio/CG9754 connects the Piwi-piRNA complex to the cellular heterochromatin machinery. Genes Dev. 2015, 29, 2258–2271.
- Yu, Y.; Gu, J.; Jin, Y.; Luo, Y.; Preall, J.B.; Ma, J.; Czech, B.; Hannon, G.J. Panoramix enforces piRNA-dependent cotranscriptional silencing. Science 2015, 350, 339–342.
- Iwasaki, Y.W.; Murano, K.; Ishizu, H.; Shibuya, A.; Iyoda, Y.; Siomi, M.C.; Siomi, H.; Saito, K. Piwi Modulates Chromatin Accessibility by Regulating Multiple Factors Including Histone H1 to Repress Transposons. Mol. Cell 2016, 63, 408–419.
- Klenov, M.; Lavrov, S.A.; Korbut, A.P.; Stolyarenko, A.D.; Yakushev, E.Y.; Reuter, M.; Pillai, R.S.; Gvozdev, V.A. Impact of nuclear Piwi elimination on chromatin state in Drosophila melanogaster ovaries. Nucleic Acids Res. 2014, 42, 6208–6218.
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Tóth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013, 27, 390–399.
- Gu, W.; Shirayama, M.; Conte, D.; Vasale, J.; Batista, P.J.; Claycomb, J.M.; Moresco, J.J.; Youngman, E.M.; Keys, J.; Stoltz, M.J.; et al. Distinct Argonaute-Mediated 22G-RNA Pathways Direct Genome Surveillance in the C. elegans Germline. Mol. Cell 2009, 36, 231–244.
- Lee, H.-C.; Gu, W.; Shirayama, M.; Youngman, E.; Conte, D.; Mello, C.C. C. elegans piRNAs Mediate the Genome-wide Surveillance of Germline Transcripts. Cell 2012, 150, 78–87.
- Akay, A.; Di Domenico, T.; Suen, K.M.; Nabih, A.; Parada, G.E.; Larance, M.; Medhi, R.; Berkyurek, A.C.; Zhang, X.; Wedeles, C.J.; et al. The Helicase Aquarius/EMB-4 Is Required to Overcome Intronic Barriers to Allow Nuclear RNAi Pathways to Heritably Silence Transcription. Dev. Cell 2017, 42, 241–255.e8.
- Luteijn, M.J.; Van Bergeijk, P.; Kaaij, L.J.T.; Almeida, M.V.; Roovers, E.F.; Berezikov, E.; Ketting, R.F. Extremely stable Piwi-induced gene silencing inCaenorhabditis elegans. EMBO J. 2012, 31, 3422–3430.
- Shen, E.-Z.; Chen, H.; Ozturk, A.R.; Tu, S.; Shirayama, M.; Tang, W.; Ding, Y.-H.; Dai, S.-Y.; Weng, Z.; Mello, C.C. Identification of piRNA Binding Sites Reveals the Argonaute Regulatory Landscape of the C. elegans Germline. Cell 2018, 172, 937–951.e18.
- Cornes, E.; Bourdon, L.; Singh, M.; Mueller, F.; Quarato, P.; Wernersson, E.; Bienko, M.; Li, B.; Cecere, G. piRNAs initiate transcriptional silencing of spermatogenic genes during C. elegans germline development. Dev. Cell 2021, 57, 180–196.e7.
- Castanotto, D.; Tommasi, S.; Li, M.; Li, H.; Yanow, S.; Pfeifer, G.P.; Rossi, J.J. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol. Ther. 2005, 12, 179–183.
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small Interfering RNA-Induced Transcriptional Gene Silencing in Human Cells. Science 2004, 305, 1289–1292.
- Suzuki, K.; Shijuuku, T.; Fukamachi, T.; Zaunders, J.; Guillemin, G.; Cooper, D.; Kelleher, A. Prolonged transcriptional si-lencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J. RNAi Gene Silenc. Int. J. RNA Gene Target. Res. 2005, 1, 66–78.
- Ting, A.H.; Schuebel, K.E.; Herman, J.G.; Baylin, S.B. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat. Genet. 2005, 37, 906–910.
- Park, C.W.; Chen, Z.; Kren, B.T.; Steer, C.J. Double-stranded siRNA targeted to the huntingtin gene does not induce DNA methylation. Biochem. Biophys. Res. Commun. 2004, 323, 275–280.
- Janowski, B.A.; Huffman, K.E.; Schwartz, J.; Ram, R.; Hardy, D.; Shames, D.S.; Minna, J.D.; Corey, D.R. Inhibiting gene expression at transcription start sites in chromosomal DNA with antigene RNAs. Nat. Chem. Biol. 2005, 1, 216–222.
- Weinberg, M.S.; Villeneuve, L.M.; Ehsani, A.; Amarzguioui, M.; Aagaard, L.; Chen, Z.-X.; Riggs, A.D.; Rossi, J.J.; Morris, K.V. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 2005, 12, 256–262.
- Kim, D.H.; Villeneuve, L.M.; Morris, K.V.; Rossi, J.J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797.
- Maniataki, E.; Mourelatos, Z. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev. 2005, 19, 2979–2990.
- Cho, S.; Park, J.S.; Kang, Y.-K. AGO2 and SETDB1 cooperate in promoter-targeted transcriptional silencing of the androgen receptor gene. Nucleic Acids Res. 2014, 42, 13545–13556.
- Kim, D.H.; Saetrom, P.; Snøve, O., Jr.; Rossi, J.J. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235.
- Ittmann, M.M. Cell cycle control of the BN51 cell cycle gene which encodes a subunit of RNA polymerase III. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1994, 5, 783–788.
- Younger, S.T.; Corey, D.R. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Res. 2011, 39, 5682–5691.
- Liu, J.; Liu, Z.; Corey, D.R. The Requirement for GW182 Scaffolding Protein Depends on Whether Argonaute Is Mediating Translation, Transcription, or Splicing. Biochemistry 2018, 57, 5247–5256.
- DI Mauro, V.; Crasto, S.; Colombo, F.S.; Di Pasquale, E.; Catalucci, D. Wnt signalling mediates miR-133a nuclear re-localization for the transcriptional control of Dnmt3b in cardiac cells. Sci. Rep. 2019, 9, 9320.
- Zheng, L.; Chen, Y.; Ye, L.; Jiao, W.; Qiangsong, T.; Mei, H.; Li, D.; Yang, F.; Li, H.; Huang, K.; et al. miRNA-584-3p inhibits gastric cancer progression by repressing Yin Yang 1- facilitated MMP-14 expression. Sci. Rep. 2017, 7, 8967.
- Seila, A.C.; Calabrese, J.M.; Levine, S.S.; Yeo, G.W.; Rahl, P.B.; Flynn, R.A.; Young, R.A.; Sharp, P.A. Divergent Transcription from Active Promoters. Science 2008, 322, 1849–1851.
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484–1488.
- Portnoy, V.; Huang, V.; Place, R.F.; Li, L.-C. Small RNA and transcriptional upregulation. Wiley Interdiscip. Rev. RNA 2011, 2, 748–760.
- Younger, S.T.; Pertsemlidis, A.; Corey, D.R. Predicting potential miRNA target sites within gene promoters. Bioorganic Med. Chem. Lett. 2009, 19, 3791–3794.
- Place, R.F.; Li, L.-C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613, Correction in Proc. Natl. Acad. Sci. USA 2018, 115, E3325.
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.-C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2011, 40, 1695–1707.
- Song, Y.; Zhao, C.; Dong, L.; Fu, M.; Xue, L.; Huang, Z.; Tong, T.; Zhou, Z.; Chen, A.; Yang, Z.; et al. Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis 2007, 29, 307–315.
- Aaltonen, K.; Amini, R.-M.; Heikkilä, P.; Aittomäki, K.; Tamminen, A.; Nevanlinna, H.; Blomqvist, C. High cyclin B1 expression is associated with poor survival in breast cancer. Br. J. Cancer 2009, 100, 1055–1060.
- Majid, S.; Dar, A.A.; Saini, S.; Yamamura, S.; Hirata, H.; Tanaka, Y.; Deng, G.; Dahiya, R. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer 2010, 116, 5637–5649.
- Qu, H.; Zheng, L.; Pu, J.; Mei, H.; Xiang, X.; Zhao, X.; Li, D.; Li, S.; Mao, L.; Huang, K.; et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Hum. Mol. Genet. 2015, 24, 2539–2551.
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334.
- Portnoy, V.; Lin, S.H.S.; Li, K.H.; Burlingame, A.; Hu, Z.-H.; Li, H.; Li, L.-C. saRNA-guided Ago2 targets the RITA complex to promoters to stimulate transcription. Cell Res. 2016, 26, 320–335.
- Jain, A.; Bacolla, A.; Chakraborty, P.; Grosse, F.; Vasquez, K.M. Human DHX9 Helicase Unwinds Triple-Helical DNA Struc-tures. Biochemistry 2010, 49, 6992–6999.
- Marton, H.A.; Desiderio, S. The Paf1 complex promotes displacement of histones upon rapid induction of transcription by RNA polymerase II. BMC Mol. Biol. 2008, 9, 4.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
821
Revisions:
3 times
(View History)
Update Date:
07 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No