Lysophosphatidic acid (LPA) is a growth factor-like lipid mediator that regulates various physiological functions via activation of multiple LPA G protein-coupled receptors. Mitochondria have been suggested to be the primary origin of oxidative stress via the overproduction of reactive oxygen species (ROS). Mitochondria are responsible for producing ATP through oxidative phosphorylation (OXPHOS) and have a calcium buffering capacity for the cell. Defects in mitochondria will lead to declined antioxidant capacity and cell apoptosis. siRNA-mediated depletion of LPA3 leads to the depolarization of mitochondrial potential (ΔΨm) and cellular ROS accumulation. In addition, the depletion of LPA3 enhances cisplatin-induced cytochrome C releasing. This indicates that LPA3 is essential to suppress the mitochondrial apoptosis pathway. LPA3 is also shown to improve mitochondrial ADP-ATP exchange by enhancing the protein level of ANT2. On the other hand, LPA3 regulates calcium uptake from the ER to mitochondria via the IP3R1-VDAC1 channel. Moreover, activation of LPA3 by selective agonist OMPT rescues mitochondrial homeostasis of H2O2-induced oxidative stress cells and HGPS patient fibroblasts by improving mitochondrial ΔΨm and OXPHOS.
1. Introduction
Lysophosphatidic acid (1- or 2-acyl-sn-glycerol 3-phosphate/radyl-glycerol-phosphate, LPA) is a bioactive lipid mediator produced both intracellularly and extracellularly from membrane phospholipids and is detected robustly in all eukaryotic tissues and blood plasma
[1]. LPA evokes various cellular responses by activating six distinct G protein-coupled receptors (GPCRs) localized in the plasma membrane of various cells, mediating cell proliferation, migration, and cytoskeletal reorganization
[2]. These receptors are subdivided into the endothelial cell differentiation gene family (Edg, LPA
1-LPA
3)
[3] and the P2Y purinergic receptor family (LPA
4-LPA
6)
[4]. Upon binding with LPA, conformational changes of LPA receptors allow them to act as guanine nucleotide exchange factors for one or more of the four classes of heterotrimeric G proteins, G
12/13, G
q/11, G
i/o, and G
s, which initiate signaling cascades through downstream molecules such as Rho, IP3-DAG, adenylyl cyclase and Ras/mitogen-activated protein kinase (MAPK) pathways
[5][6][7]. Studies have documented the wide range of LPA downstream signaling pathways involving diverse physiological and pathophysiological functions, including nervous/vascular system development, reproduction, angiogenesis, cancer progression, and hematopoiesis
[8][9][10].
Moreover, researchers also showed that LPA can exert different functions according to its GPCRs expression pattern. For instance, researchers' recent studies suggest that LPA
2 and LPA
3 expressed at different stages of hematopoietic progenitors decide the fate of blood cell differentiation
[11][12]. In addition, studies proposed the concept that the profile of LPA receptors change during the cell aging process which affect activities of the downstream pathway
[13][14]. Similarly, researchers also observed that LPA
3 decreases in the senescent and premature aging disease, Hutchinson-Gilford progeria symptom (HGPS) model
[11][15]. LPA
3 knockout zebrafish larvae researchers established showed senescent phenotypes and lower locator activity, and the adult fish have a shorter life span than wild-type zebrafish
[16]. LPA also protects cells from mitochondrial apoptosis by enhancing JNK-mediated phosphorylation of Bcl-2 and ERK1/2 pathways and by accumulating the anti-apoptotic protein, Mcl-1
[17][18]. These beneficial effects of LPA on mitochondria can align with researchers' findings that activation of LPA
3 in HGPS cells rescued premature aging phenotype. Notably, researchers' recently published studies showed that activation of LPA
3 stabilizes the protein of nuclear factor erythroid 2-related factor (Nrf2), consequently promoting the expression of antioxidants and reducing mitochondrial superoxide
[15]. This evidence suggests that the potentiation of LPA
3 signaling may constitute a new approach against aging progress and mitochondrial oxidative stress.
Mitochondria are currently considered a critical component of intracellular signaling, immune response, and a modulator of cell replication, more than simple bioenergetic factories. The development of a wide range of aging-related diseases is highly associated with the decline of mitochondrial quality and activity. It is widely accepted that, during the aging process, the free radical (reactive oxygen species, ROS) leads to cumulative damage and irreversible cell damage. The mitochondria are considered the central regulators of redox activities and oxidative stress, called mitochondrial ROS (mtROS). Therefore, mitochondrial dysfunctions have been suggested to be the hallmark and driver of aging
[19]. In addition, HGPS, which is severe premature aging, shows similar signaling pathway patterns as normal aging cells
[20]. This syndrome is typically caused by a silent mutation (c. 1824C>T; p. Gly608Gly) of LMNA that activates an alternative pre-mRNA cryptic splicing site and causes a 150-nucleotide, 50 amino acid deletion. The missing sequence includes a recognition site for ZMPSTE24 endoprotease, leading to the accumulation of an un-cleaved Lamin A isoform, named progerin
[21]. HGPS cells display significant dysregulation of oxidative phosphorylation (OXPHOS), mitochondrial activities, and ROS scavenging
[22][23]. Treatments aimed to improve the mitochondrial homeostasis were shown to ameliorate the cell senescence of HGPS cells
[24].
2. LPA3 Signaling Is Crucial to Maintain Mitochondria Homeostasis and ROS Accumulation
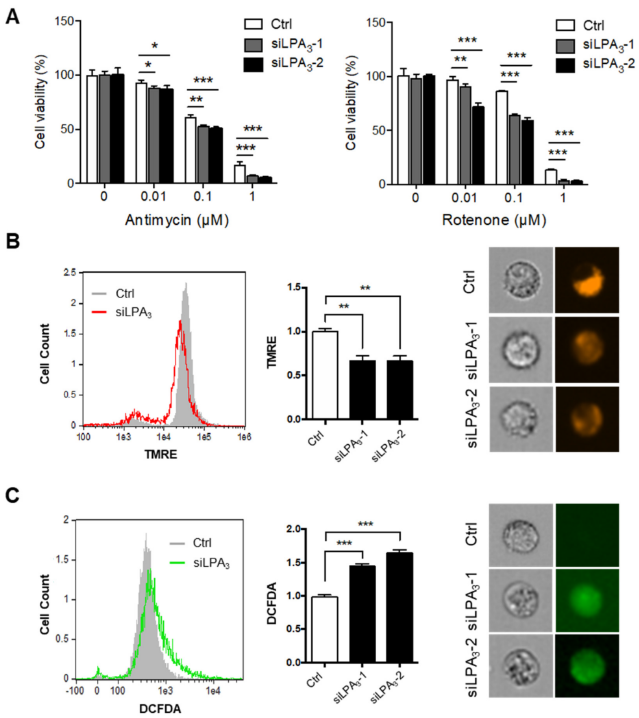
To delineate the role of LPA3 and its underlying mechanism in regulating mitochondrial activity, researchers subjected the siRNA control and LPA3 siRNA-treated human cervical cancer HeLa cells to a cell proliferation assay with electron transport chain (ETC) complex I inhibitor, Rotenone, and complex III inhibitor, Antimycin A (Figure 1A). Two LPA3 siRNA were used in this entry. LPA3 siRNA knockdown cells were susceptible to the two ETC inhibitors, suggesting that that LPA3 was required to counter mitochondrial stress. Furthermore, the mitochondrial membrane potential (ΔΨm) was measured by tetramethylrhodamine ethyl ester, perchlorate (TMRE) staining. TMRE is a cell-permeant that shows the level of negative charge across the healthy inner mitochondrial membrane to the matrix. ETC complex I, III, and IV serve as proton pumps to generate a proton gradient (ΔΨm) across the inner mitochondrial membrane as energy storage for driving OXPHOS. Thus, mitochondrial ΔΨm is a remarkable indicator for mitochondrial healthiness. Researchers found significantly depolarized mitochondrial ΔΨm in LPA3-depleted cells (Figure 1B). Since superoxide can leak out from mitochondria to contribute endogenous ROS, the cellular ROS was detected by 2′,7′–dichlorofluorescein diacetate (DCFDA) staining. It showed that the cellular ROS accumulation increased in LPA3-depleted cells (Figure 1C).
Figure 1. Impairment of mitochondrial homeostasis in LPA3–deficient cells. (A) Control (siRNA control) and two LPA3 siRNA knockdown HeLa cells were subjected to a cell proliferation assay against mitochondrial electron transport chain (ETC) inhibitors, complex-III inhibitor Antimycin A and complex-I inhibitor Rotenone, at indicated doses for 72 h. LPA3 siRNA knockdown cells were sensitive to inhibitors against ETC. (B) Mitochondrial ΔΨm of siRNA control and two LPA3 siRNA knockdown cells were analyzed with TMRE dye by imaging flow cytometry. TMRE fluorescence was reduced in two LPA3 siRNA knockdown cells. Representative images of TMRE stained cells were shown in the right panel. (C) Cellular ROS of siRNA control and two LPA3 siRNA knockdown cells were analyzed by DCFDA dye. Cells were stained with 20 μM of DCFDA at 37 °C for 30 min, followed by imaging flow cytometry analysis. DCFDA fluorescence showed increasing in two LPA3 siRNA knockdown cells. Representative images of DCFDA stained cells were shown in the right panel. The bar graphs were generated from three independent analyses. * p < 0.05; ** p < 0.01; *** p < 0.001.
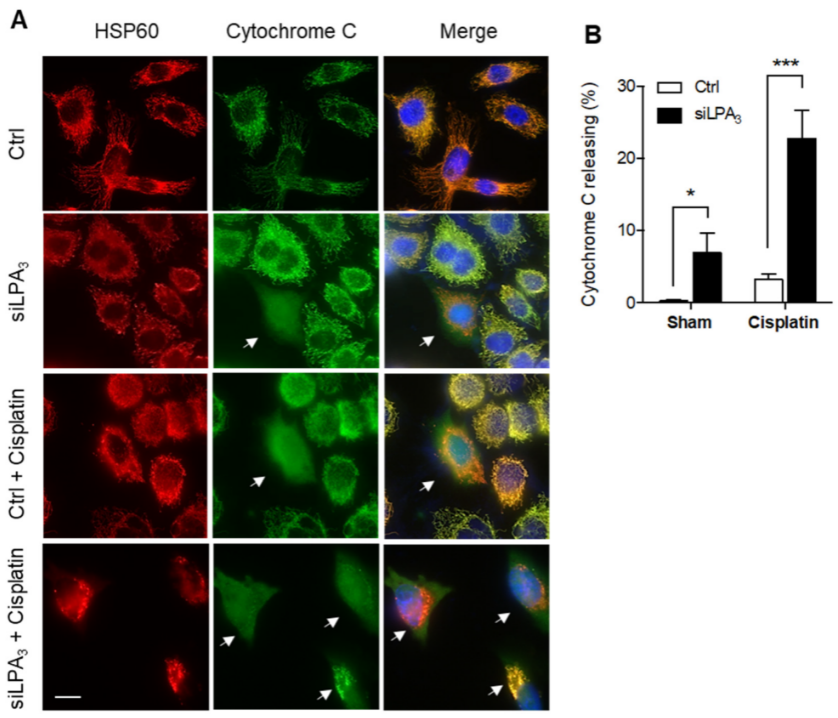
Moreover, cytochrome
c release was monitored to detect the healthiness of mitochondria. In healthy mitochondria, cytochrome
c locates at the mitochondrial intermembrane space and functions as an electron shuttle in the ETC. The mitochondrial damage induces the outer membrane’s permeabilization, facilitating cytochrome
c releasing from the mitochondria to cytosol. The released cytochrome
c in the cytosol further mediates the activation of caspase-3 dependent apoptosis
[25]. LPA
3 siRNA knockdown cells displayed an increased ratio of cytochrome
c release. Moreover, cisplatin as a mitochondrial DNA damaging reagent was treated to induce cytochrome
c releasing
[26]. Relative to siRNA control HeLa cells, LPA
3 depletion cells were more sensitive to cisplatin-induced cytochrome
c releasing (
Figure 2). These results highlight the crucial role of LPA
3 in mitochondria homeostasis, evidenced by the cytochrome
c releasing, mitochondrial ΔΨm depolarization, and cellular ROS accumulation in LPA
3-depletion cells.
Figure 2. Knockdown of LPA3 increased the release of cytochrome c from mitochondria to the cytosol. (A) Representative images of immunofluorescent staining that showed cytochrome c (green) and mitochondria marker HSP60 (red) in siRNA control (Ctrl) and LPA3 siRNA knockdown (siLPA3) HeLa cells. Nuclei were stained with DAPI. Cytochrome c releasing cells were indicated with the white arrow. Scale bar represents 10 µm. (B) Knockdown of LPA3 increased the release of cytochrome c from mitochondria to the cytosol and enhanced cellular sensitivity to 0.5 μM of cisplatin-induced cytochrome c releasing. The bar graphs were generated from three independent analyses. * p < 0.05; *** p < 0.001.
3. LPA3 Signaling Is Involved in Mitochondrial ADP-ATP Exchange
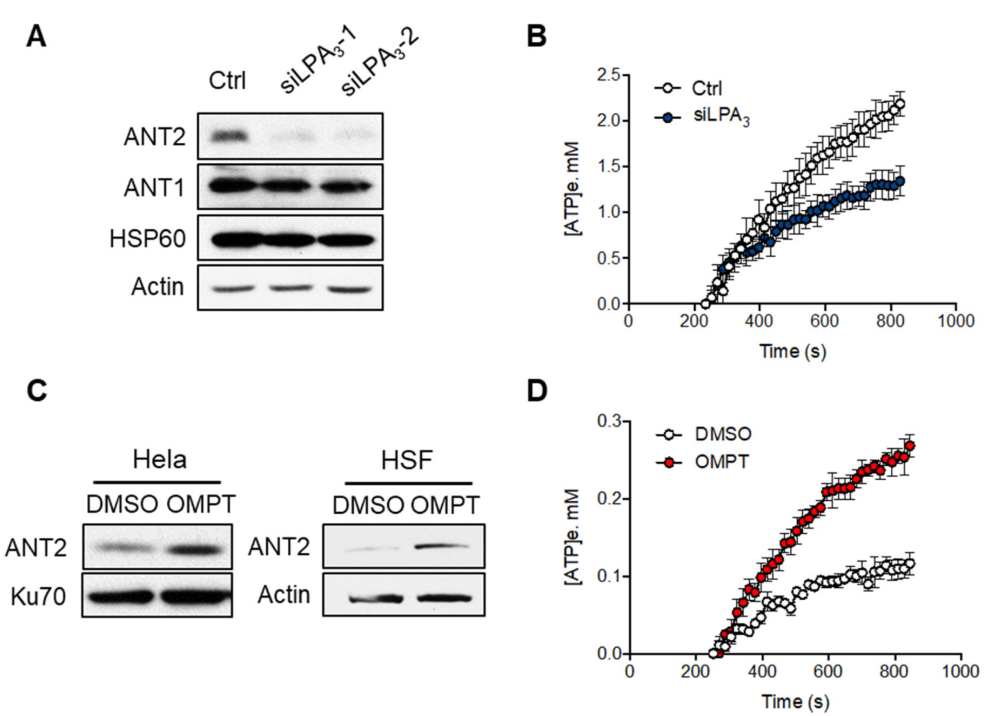
Previous research has shown that low expression of ANT2 is linked to mitochondrialinherent to cell senescence
[27]. Therefore, researchers aimed to test if LPA
3 signaling regulates protein level and activity of ANT2. The Western blot analysis of mitochondrial proteins showed that the protein level of ANT2 was significantly reduced by siLPA
3, whereas ANT1 and the mitochondrial matrix chaperone HSP60 were not (
Figure 3A). Adenine nucleotide translocase 2 (ANT2) is the crucial regulator for mitochondrial homeostasis by exchanging ADP and ATP across the mitochondrial inner membrane. In most physiological conditions, electron transport through the ETC is tightly coupled with the phosphorylation of ADP to ATP by F0F1-ATPase. Impairing the ADP supply to mitochondria will slow down OXPHOS, increase the redox status of mitochondria, and thereby drive excess ROS production
[28]. Thus, researchers measured the mitochondrial ADP-ATP exchange using an Mg
2+ sensitive fluorescent probe Magnesium Green 5K+ (abbreviated as MgGreen)
[29][30]. Since biologically active ATP and ADP are bound with Mg
2+ and have different affinity (K
ADP = 0.906 mM and K
ATP = 0.114 mM). The dynamics of free extra-mitochondrial Mg
2+ can be measured and determine ADP-ATP exchange activity in cells. Researchers observed that the LPA
3 knockdown cells by siRNA showed a significant attenuation in ADP-ATP exchange (
Figure 3B). Moreover, activation of LPA
3 with its highly selective agonist, OMPT, increased the ANT2 protein expression in both Hela and human skin fibroblast (HSF) cells (
Figure 3C) and the activity of ADP-ATP exchange (
Figure 3D). These results suggest that LPA
3 signaling is crucial to regulate the mitochondrial ADP-ATP exchange.
Figure 3. LPA3 modulated mitochondrial ADP-ATP exchange activity. (A) Representative western blot analysis of the attenuation of ANT2 protein level, but not ANT1, in LPA3 knockdown cells. Actin was used as a loading control. (B) siRNA control (Ctrl) and LPA3 siRNA knockdown (siLPA3) HeLa cells were subjected to ADP-ATP exchange assay using MgGreen 5K+ fluorescent dye. The exchange was stimulated by 5 mM of ADP. MgGreen 5K+ fluorescence was first measured to indicate changes in free magnesium concentration after adding ADP. The decreased free magnesium concentration was then converted to concentration of efflux ATP ([ATP]e, mM). Treatment of LPA3 siRNA attenuated the ADP-ATP exchange. (C) Representative western blot analysis of the induction of ANT2 protein level by treatment of 50 nM of LPA3 agonist OMPT for 48 h in HeLa and Human skin fibroblast (HSF). (D) Treatment with 50 nM of OMPT increased the ADP-ATP exchange. The graphs were generated from three independent analyses.
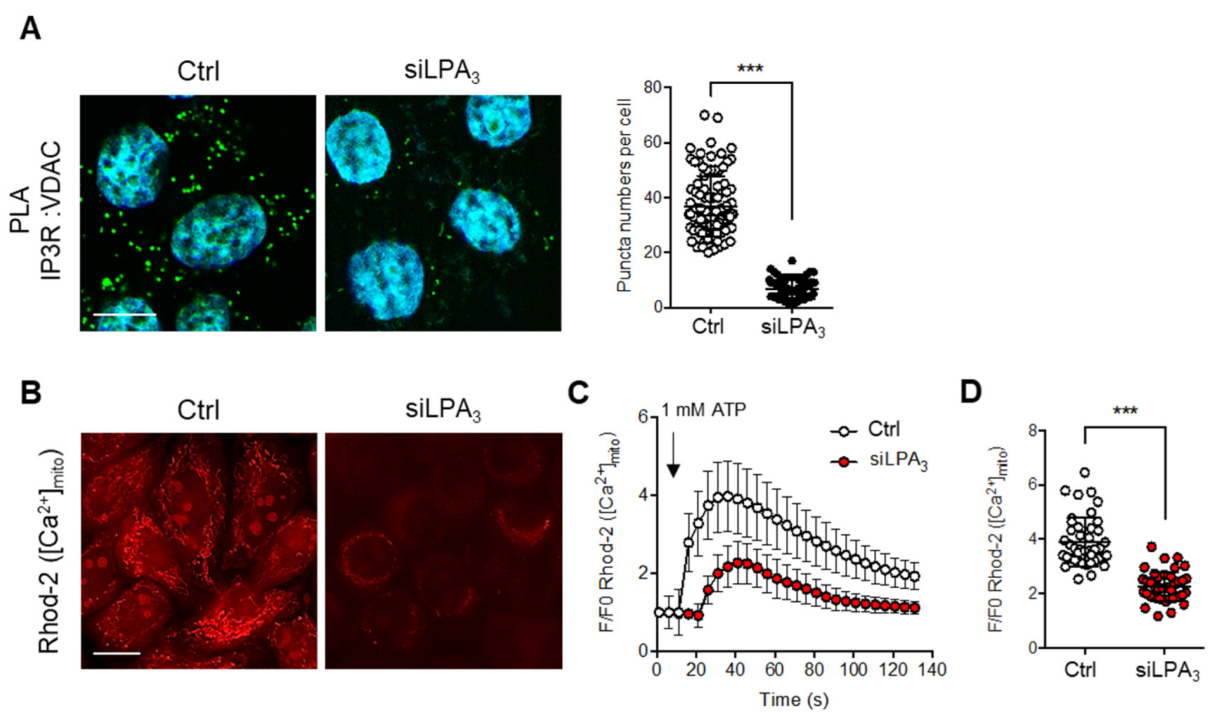
4. LPA3 Signaling Is Involved in the Regulation of Mitochondrial Ca2+ Influx from ER to Mitochondria
On the other hand, the mitochondrial electrochemical proton gradient (ΔμH
+) is the major component of the mitochondria ΔΨ. ΔμH
+ drives the flow of H
+ through ATP synthase in a reaction coupled to the generation of ATP from ADP
[31] and represents a substantial driving force for Ca
2+ accumulation
[32]. Mitochondrial Ca
2+ traffics from ER to mitochondria in response to physiological stimuli and stresses
[33][34]. The protein channel complex IP3R1-VDAC1 was suggested to transfer the mitochondrial Ca
2+ influx from the ER to mitochondrial intermembrane space. Therefore, the interacting structure of IP3R1-VDAC1 was detected with in situ proximity ligation assay (PLA) by probing IP3R1 and VDAC1 antibodies. The hybridization with PLA secondary antibodies coupled to fluorescent oligonucleotides hybridizes if the distance is <40 nm
[35]. The interactions between IP3R1-VDAC1 were decreased in LPA
3 depleted cells (
Figure 4A). To verify the Ca
2+ transport efficiency from the ER to mitochondria, the mitochondrial Ca
2+ was measured by mitochondria-specific Ca
2+ indicator Rhod-2 AM with live cell image analysis. ATP was used to stimulate P2Y receptors-dependent intracellular Ca
2+ releasing from the ER. By measuring the dynamics and the peaks of Rhod-2 AM fluorescence, researchers demonstrated that the rate and amount of Ca
2+ efflux into mitochondria following administration of ATP treatment were significantly attenuated in LPA
3 depleted cells (
Figure 4B–D). The results highlight that LPA
3 signaling is important to support the structure of the Ca
2+ channel complex and Ca
2+ trafficking into the mitochondria from ER.
Figure 4. LPA3 ablation reduced IP3R1 and VDAC1 association and disrupts mitochondria Ca2+ influx. (A) Representative images of in situ proximity ligation assay were used to unveil the IP3R1-VDAC1 interaction in siRNA control (Ctrl) and LPA3 siRNA knockdown (siLPA3) HeLa cells. Green dots indicated the location of the interaction. Nuclei were stained with DAPI. The association between VDAC1 and IP3R1 was significantly attenuated in LPA3 knockdown cells. Scale bar represents 10 µm. (B) Representative images of mitochondrial [Ca2+] analysis by mitochondria-specific Ca2+ indicator Rhod-2 AM. Mitochondria [Ca2+] were measured in siRNA control (Ctrl) and LPA3 siRNA knockdown (siLPA3) HeLa cells. Scale bar represents 10 µm. (C) Representative traces of intramitochondrial Ca2+ dynamic. 1 mM of ATP was used to stimulate P2Y receptors-dependent intracellular Ca2+ releasing from ER. (D) The average Rhod-2 AM peak fluorescence following administration of ATP was decreased in LPA3 knockdown cells. The graphs were generated from three independent analyses. *** p < 0.001.
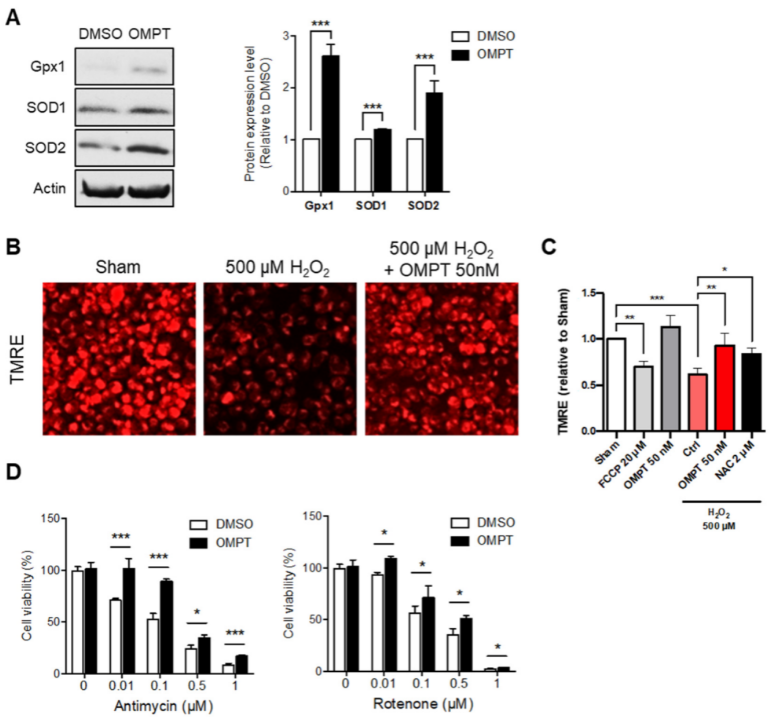
5. LPA3 Signaling Participates in the Regulation of Mitochondrial Stress Response
Since researchers have shown that LPA
3 plays a crucial role as regulator of mitochondria, researchers next aimed to discuss whether LPA
3 signaling may constitute a new approach against oxidative stress-induced mitochondrial dysfunction. Activation of LPA
3 with its highly selective agonist, OMPT, showed potent induction of mitochondrial antioxidants, glutathione peroxidase 1 (Gpx1), and superoxide dismutase type 1 and type 2 (SOD1 and SOD2) protein expression in human cervical cancer HeLa cells (
Figure 5A). To further delineate the role of LPA
3 in regulating the mitochondrial oxidative stress response, researchers subjected HeLa cells with treatment of hydrogen peroxide (H
2O
2), followed by analysis of the mitochondrial membrane potential (ΔΨm) by TMRE staining. Researchers found significantly depolarized mitochondrial ΔΨm in H
2O
2-induced oxidative stress and rescued with OMPT treatment (
Figure 5B,C). The mitochondrial uncoupler carbonyl cyanide
p-trifluoromethoxyphenylhydrazone (FCCP) was used as a negative control to depolarize mitochondria. The antioxidant N-acetylcysteine (NAC) was used as the positive control to rescue ΔΨm. Human erythroleukemic K562 cells were also used to confirm the protection effect of OMPT. The results also suggested similar antioxidant function of OMPT. In addition, researchers further subjected HeLa cells to a cell proliferation assay with inhibitors against OXPHOS. The respiratory complex I (NADH: ubiquinone oxidoreductase) and respiratory complex III (ubiquinone: cytochrome
c reductase) are recognized to produce mtROS relevant to pathological aged diseases
[36]. While highly mtROS is presented, the respiratory complexes are likely to be attacked and drive reverse electron transfer (RET), which at the same time leads to electron leak to damage cell compartments
[37]. The OMPT treated cells were highly resistant to ETC inhibitors Rotenone (complex I) and Antimycin A (complex III) induced oxidative stress (
Figure 5D). These findings suggested the protective functions of LPA
3 to alleviate mitochondrial function during oxidative stress.
Figure 5. LPA3 protected against oxidative stress-induced mitochondria dysfunction. (A) Western blot results show that activating LPA3 by 50 nM LPA3 agonist, OMPT for 7 h enhanced protein levels of Gpx1, SOD1, and SOD2 in HeLa cell. Actin was used as a loading control. (B) Representative images of mitochondrial membrane potential (ΔΨm) analysis by TMRE staining. Scale bar represents 10 µm. (C) Cells were pre-treated 50 nM of OMPT or 2 μM of NAC for 4 h respectively, followed by 3 h 500 μM of H2O2 treatment. Cells were further stained with 200 nM of TMRE and 1 μM of DAPI for 30 min at 37 °C, followed by Imaging Plate Reader analysis. The fluorescent intensity of DAPI was used as cell number control. HeLa cells showed decreased mitochondria ΔΨm under 500 μM of H2O2 treatment. 50 nM of OMPT and 2 μM of NAC protected cells against H2O2-induced mitochondrial ΔΨm loss. FCCP was used as a positive control for loss of mitochondrial ΔΨm. (D) HeLa cells were subjected to a cell proliferation assay against mitochondrial electron transport chain (ETC) inhibitors, complex-III inhibitor Antimycin A (left), and complex-I inhibitor Rotenone (right), at indicated doses for 72 h. Cells showed protective effects against ETC inhibitors in 50 nM of OMPT treatment. The bar graphs were generated from three independent analyses. * p < 0.05; ** p < 0.01; *** p < 0.001.
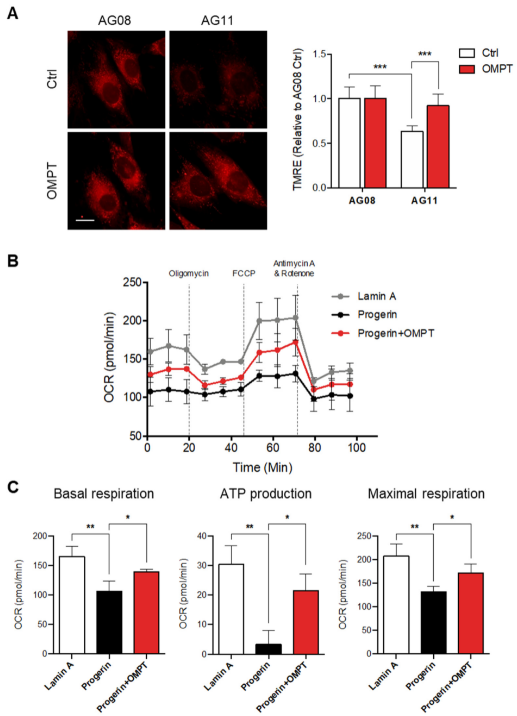
6. LPA3 Activation Rescues Mitochondrial Activity in Hutchinson-Gilford Progeria Syndrome Cells
The ROS leads to cumulative damage during the aging process and results in irreversible cell damage. The mitochondria are considered the central regulators of redox activities, and oxidative stress is referred to as mitochondrial ROS (mtROS). Therefore, the alterations in mitochondrial functions have been suggested to be the hallmark and the drivers of aging. Researchers' previous publication indicated that the expression level of LPA
3 declined in the aging disease Hutchinson-Gilford progeria syndrome (HGPS). Enhancement of LPA
3 signaling by agonist is suggested to ameliorate premature aging
[11][15]. HGPS also displayed the dysregulation of OXPHOS and mitochondrial activity
[22]. Therefore, researchers aimed to explore whether the protective effects of LPA
3 on mitochondria and against oxidative stress can constitute a new approach to HGPS treatment. Researchers first showed that OMPT treatment protected normal fibroblast against H
2O
2 induced oxidative stress. Next, researchers analyzed the mitochondrial ΔΨm in HGPS patient fibroblast AG11 and age-matched control fibroblast line AG08 cells by TMRE staining. Relative to the control AG08 cells, HGPS AG11 cells displayed depolarized mitochondrial ΔΨm. Activation of LPA
3 signaling had no effect on control cells but rescued depolarized mitochondrial ΔΨm in HGPS cells (
Figure 6A). Moreover, researchers subjected lamin A control and HGPS progerin expression HEK 293 cells to the Agilent Seahorse XFe24 analyzer to determine mitochondrial OXPHOS by measuring the oxygen consumption rate (OCR). The OCR was determined under basal conditions followed by the addition of oligomycin (ATP synthase inhibitor), FCCP (mitochondrial uncoupler), and rotenone plus antimycin A (respiratory complex I and III inhibitors) to estimate individual parameters of mitochondria. Seahorse XF Cell Mito Stress assays revealed that the OXPHOS was significantly disrupted in HGPS cells (
Figure 6B,C). OMPT treatment rescued the OCR level of basal respiration, ATP production, and maximal respiration (
Figure 6B,C). Together, researchers conclude that LPA
3 acts as the upstream signal to maintain mitochondria homeostasis through regulating mitochondria Ca
2+ trafficking and ADP-ATP exchange. Loss of LPA
3 leads to dysregulation of mitochondria biogenesis, OXPHOS, and ROS control in HGPS and aging cells. Activating LPA
3 signaling by its agonist OMPT can rescue the mitochondrial defects from these pathological conditions. These results suggested that the potentiation of LPA
3 signaling may constitute a new approach against aging progress and mitochondrial oxidative stress.
Figure 6. Activation of LPA3 rescued mitochondrial activity in Hutchinson-Gilford progeria syndrome cells. (A) HGPS patient fibroblast AG11 and age-matched control fibroblast line AG08 cells were treated with 50 nM of OMPT or DMSO control for 48 h. Cells were further stained with 200 nM of TMRE for 30 min at 37 °C, followed by Imaging analysis. Analysis of mitochondrial ΔΨm by TMRE staining showed decreased mitochondrial ΔΨm in HGPS AG11 cells relative to the AG08 control cells. OMPT increased the mitochondrial ΔΨm in HGPS AG11, but not AG08 control cells. Representative images of TMRE stained cells were shown in the left panel. Scale bar represents 10 µm. (B) Lamin A control and HGPS progerin expression HEK 293 cells were subjected to Agilent Seahorse XFe24 and analyzed to measure oxidative phosphorylation (OXPHOS) by measuring the oxygen consumption rate (OCR). (C) HGPS cells displayed a reduction of OCR, basal respiration, ATP production, and maximal respiration and were rescued by 50 nM of OMPT treatment. The bar graphs were generated from three independent analyses. * p < 0.05; ** p < 0.01; *** p < 0.001.
 +1 credit
+1 credit