Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Da-Wei Lin | -- | 4309 | 2022-03-30 18:41:50 | | | |
| 2 | Amina Yu | -13 word(s) | 4296 | 2022-03-31 04:54:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lin, D.; Chang, C.; Hsu, Y.; Lin, C. Glomerular Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/21183 (accessed on 09 August 2026).
Lin D, Chang C, Hsu Y, Lin C. Glomerular Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/21183. Accessed August 09, 2026.
Lin, Da-Wei, Cheng-Chih Chang, Yung-Chien Hsu, Chun-Liang Lin. "Glomerular Diseases" Encyclopedia, https://encyclopedia.pub/entry/21183 (accessed August 09, 2026).
Lin, D., Chang, C., Hsu, Y., & Lin, C. (2022, March 30). Glomerular Diseases. In Encyclopedia. https://encyclopedia.pub/entry/21183
Lin, Da-Wei, et al. "Glomerular Diseases." Encyclopedia. Web. 30 March, 2022.
Copy Citation
Glomerular disease is a disease of glomerular inflammation caused by immune-mediated damage to capillary endothelium, mesangium, or basement membrane. The clinical presentations of glomerular disease are diverse. Patients may have asymptomatic microscopic or gross hematuria and often develop proteinuria and even the severe as nephrotic syndrome.
minimal change disease
IgA nephropathy
membranous nephropathy
1. Minimal Change Disease and Primary Focal Segmental Glomerulosclerosis
Minimal change disease is the most common cause of the nephrotic syndrome. It frequently occurs in children older than 1 year old, accounting for 70–90%. This disease is often associated with dyslipidemia and hypercoagulability. After puberty, the incidence decreases significantly [1][2]. This disease is named by the absence of significant findings in glomeruli under the light microscope. Only foot process effacement by the electron microscope is noticed, without depositions of antibodies. It was showed that focal segmental glomerulosclerosis and are termed accordingly by that finding [3]. These pathological findings are denoted as podocytopathy, which includes diffuse mesangial sclerosis (DMS), minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), and collapsing glomerulopathy [4]. Minimal change disease and primary focal segmental glomerulosclerosis are thought of as a continuum of the same disease-podocytopathy. Evolution from minimal change disease to primary focal segmental glomerulosclerosis is possible over time [5][6][7][8][9]. DMS is rare, and collapsing glomerulopathy is regarded as a morphologic variant of FSGS [10].
In 1974, Shalhoub [11] hypothesized that minimal change disease was caused by systemic dysregulation of T cells, which produced circulating factors to modify podocyte structures. Dysregulation of T cells and circulating factors lead to foot process effacement and proteinuria. The T-cell-associated hypothesis is based on observations from disease remission after measles infection, response to treatment with glucocorticoid and cyclophosphamide, and remission alone with the occurrence of Hodgkin’s disease and viral upper airway infection [12][13][14][15][16][17][18]. In 2011, Shimada [19] suggested that MCD was a ‘two hits’ disorder. First, the overexpression of CD80 on podocytes, possibly mediated with Toll-like receptor 3 (TLR3), is known to be induced by allergen, cytokine, or microbial products. Podocytes then behave like antigen-presenting cells [20][21][22][23][24][25]. These overexpressed CD80s first interact with NEPH1, disrupting slit diaphragms [22]. Second, T cell response is activated while CD80 binds to its receptor CD28 on T cells. This response can be terminated by CD80 binding to another receptor cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), expressed by Treg (CD4+CD25+FoxP3+) [26][27]. Dysfunction of Treg to express CTLA-4 is the second hit [28]. Decreased expression of IL-10 by Treg, which inhibits the expression of CD80, is also noticed [29]. The binding of CD80 and CD28 in MCD increases the expression of phosphor SRC, which leads to dephosphorylation of synaptopodin and associated foot process injury [30]. Th17 is another subset of abnormally dysregulated T cells. In the animal models of adriamycin-induced nephrosis, Th17 is critically involved in the downregulation of phospho-nephrin and Bcl-2 by overexpressing c-maf inducing protein (c-mip). C-mip can inhibit Fyn-associated nephrin phosphorylation [31]. This inhibition leads to cytoskeleton disorganization with effacement of foot processes and apoptosis [32]. Besides this, protease-activated receptor-1 (PAR-1)-like mediator may exist in Th17 from MCD patients and induce a deleterious phenotypic change of podocyte via the JNK and p38 MAPK pathways [33]. A high Th17/Treg ratio is associated with increased proteinuria. Normalization in Th17/Treg ratio is seen if patients with MCD are sensitive to steroid treatment [34]. An earlier was proposed that the imbalance of Th1/Th2 cytokine activity is also related to the sensitivity to steroid treatment. Th2 activity dominance tends to be found in the steroid-sensitive primary nephrotic syndrome [35].
The implication of B cells in the pathogenesis of MCD is proposed by results of animal models and successful treatment by anti-CD20 monoclonal antibodies (for example, Rituximab) [36][37]. While histopathological in renal biopsies show no deposit of antibody or immune complex, the role of B cells in MCD is much debated. Nonetheless, B cells can also act as antigen-presenting cells to T cells and secret cytokines and chemokines to support T cell activation [38][39][40][41][42][43]. An alternative explanation is that rituximab interacts with sphingomyelin phosphodiesterase acid-like 3b protein (SMPDL) on podocytes, stabilizes the actin skeleton, and prevents apoptosis [44][45][46]. SMPDL and acid sphingomyelinase (ASM) are also expressed by Th1 [47]. Referring to the effect of rituximab (RTX) on Th17 in treating rheumatoid arthritis to decline the Th17 and suppress IL-17 expression, RTX should also play a similar role in treating MCD [48][49].
Nonetheless, autoantibodies targeting nephrin in the slit-diaphragm junctional complex in animal models have been shown to cause podocyte injury and massive proteinuria [50][51][52]. In addition, anti-nephrin autoantibodies in both animal models and cultured podocytes result in the redistribution of nephrin away from the slit diaphragm along with the separation of intercellular junctions of adjacent podocytes [50][51][53]. This redistribution of the nephrin is similar to the findings in renal biopsies of patients with nephrotic syndrome [54]. Likewise, autoantibodies targeting nephrin are discovered in a subset of patients with noncongenital childhood and adult-onset MCD [55]. These autoantibodies are reduced or absent in the circulation during the treatment response and lead to massive proteinuria recurrence in allografts of renal transplantation. Successful treatment of this recurrence by plasmapheresis/rituximab is similar to the strategy for the rapid recurrence of primary FSGS in renal transplantation [56]. This discovery of anti-nephrin autoantibodies calls attention to the heterogenic pathogenesis of MCD and supports the hypothesis of the continuum from MCD to primary FSGS.
Additionally, a few proposed circulating molecules are thought of as permeability factors, including cardiotrophin-like cytokine-1, soluble urokinase-type plasminogen activator receptor, and anti-CD40 antibody [57][58][59]. These factors lead to increase proteinuria and foot effacement. The exact roles of these circulating molecular need further investigation.
After the initial immune-mediated insults, podocytes may undergo epithelial to mesenchymal transition (EMT). In a proinflammatory milieu, transforming growth factor (TGF)-β1 is upregulated. TGF-β1 suppressed the slit diaphragm-associated protein P-cadherin, zonula occludens-1, and nephrin and induced the expression of the intermediate filament protein desmin and interstitial matrix components fibronectin and collagen I. These events increase the permeability and proteinuria production by paracellular protein flux. TGF-β1 also promotes the expression of Snail, a key transcription factor in the initiation of EMT [60].
Nephrin and NEPH1 are key proteins of the constitutive component of the slit diaphragm and are critical for podocyte stability and integrity. The spatial arrangement of the extracellular domains of nephrin and NEPH1 provide integrity of slit-diaphragm while intracellular domains play a role in signaling network for modulation of actin-cytoskeleton changes in podocytes [61]. Phosphorylation of the nephrin tyrosine residues by Src family kinases, including Src, Fyn, Lyn, and Yes, plays a critical role in stabilizing the foot process and maintaining the slit-diaphragm structure [62][63][64]. This phosphorylation induces P85/PI3K binding and recruits Nck, leading to actin reorganization [65][66]. The reduced phosphorylation level of nephrin tyrosine residues is detected in MCD and MN, and phosphorylation stabilizes and restores podocyte foot process architecture [67][68][69]. On the contrary, phosphorylation of NEPH1 tyrosine residues is found in several podocyte injury models, and this can be assumed as a therapeutic target [70][71][72]. Another molecule, ephrin-B1, at the slit-diaphragm also involves the signaling network. Phosphorylation of ephrin-B1 leads to dissociation of nephrin and Par-6 from ephrin-B1 and promotes mobility of podocytes through activation of JNK [73]. Synaptopodin and α-actinin-4 (ACTN4) are other vital proteins that regulate the actin cytoskeleton in podocytes. Synaptopodin, associated with actin, can stabilize RhoA-mediated stress fiber formation in podocytes [74]. ACTN4 cross-links filamentous actin (F-actin) and supports the podocyte structure [75]. Phosphorylation by Src kinase makes dephosphorylation of synaptopodin serine/threonine residues leads to its degradation [76], while mutation of ACTN4 is found in an autosomal dominant form of FSGS [75].
FSGS is the most common cause of end-stage renal disease among primary glomerular diseases in the united states [77]. Regarding genetic susceptibility, predisposing pathophysiological factors, and clinical courses, FSGS is categorized into five types. These include two common types (primary/idiopathic form and adaptive form) and three less common types (familial/genetic form, virus-associated form, and drug-induced form) [5]. While adaptive FSGS arises from overloaded processes involving increased single nephron GFR and intraglomerular hypertension, associated conditions include systemic hypertension, obesity, oligomeganephronia, very low birth weight, reflux nephropathy, unilateral renal agenesis, high protein diet, and any advanced renal disease with reduced functioning nephrons. Treatments for adaptive FSGS are aimed at the inhibition of the renin–angiotensin–aldosterone system to lower the glomerular filtration pressure. As human immunodeficiency virus (HIV) type 1, parvovirus B19, simian virus 40, cytomegalovirus, and Epstein–Barr virus are reported to induce virus-associated FSGS, predisposing drugs to drug-induced FSGS include heroin, interferons, lithium, pamidronate, sirolimus, calcineurin-inhibitor nephrotoxicity, and anabolic steroids. The main strategy is to stop or cure these exogenous insults for drug-induced FSGS and virus-associated FSGS. More than fifty genetic mutations expressed in podocytes or glomerular basement membranes are identified as the causes of genetic FSGS or steroid-resistant nephrotic syndrome [78][79]. These genetic/familial FSGS with single gene mutation usually express immunosuppressant resistance [80]. APOL1 risk variant associated FSGS, which is found mainly in South African ancestry, shows many different characteristics than other genetic FSGS. Presented APOL1 risk alleles confer susceptibility, but most subjects with two risk alleles may not develop kidney diseases. APOL1 risk variant associated FSGS, once diagnosed, shows rapid progression to end-stage renal disease (ESRD) [81]. Not only FSGS, these APOL1 risk variants much increase rates of hypertension-associated ESRD, HIV-associated nephropathy, end-stage of lupus nephritis, and other forms of non-diabetic kidney diseases. APOL1 associated kidney diseases may be considered as an individual entity [82].
Pediatric nephrotic syndrome is known to respond well to steroid treatment. According to the KDIGO guideline for the glomerular disease of 2021, cyclophosphamide or oral levamisole is the first-line alternative therapy for steroid-sparing regimens. Other drugs, such as mycophenolate mofetil, a calcineurin inhibitor, or rituximab, can be used as second-line treatment. Calcineurin inhibitor as the initial second-line therapy for steroid resistance nephrotic syndrome is recommended. Genetic testing to exclude congenital/familial nephrotic syndrome or genetic disorder and renal biopsy with steroid resistance, familial history of steroid-resistant nephrotic syndrome/FSGS, or syndromic features when classifications cannot be judged by clinical assessments [83]. The steroid is the first-line treatment for primary FSGS, and calcineurin inhibitor is the choice with steroid resistance. Calcineurin inhibitors can be replaced by mycophenolate combined with high-dose dexamethasone, rituximab, or ATCH if steroids are intolerant [84]. While comparing with minimal change disease, there is higher likelihood of steroid resistance and a higher rate of progression into end-stage renal disease in primary FSGS. The rate of glucocorticoid-induced remission is lower in primary FSGS. A portion of steroid-sensitive nephrotic syndrome associated with minimal change disease may become steroid resistant later as repeated renal biopsies often reveal a change of focal segmental sclerosis due to persistent and repeated podocyte injury with associated podocyte loss beyond the point of no return [9][85].
2. IgA Nephropathy
IgA nephropathy (IgAN) is the most common glomerulonephritis. The incidence of IgAN is high in Pacific-Asian regions. Familial clustering is noticed in an area where offspring often own a common ancestor [86]. Although IgAN occurs sporadically, about 5–8% of patients have relatives with biopsy-proven IgA or abnormality in urine. Patients with familial history of IgAN often have a worse prognosis [87]. However, the clinical presentations of IgA are inconsistent. Patients may be diagnosed simply due to asymptomatic hematuria or progressive renal function decline. Nephrotic range proteinuria is also commonly found. Although rapid progression to renal failure is rare, this disastrous phenotype still could happen with the crescent formation of more than 50% glomeruli [88].
The most acceptable pathogenic paradigm of IgAN is the multiple-hit pathogenesis model (Figure 1) [89]. First is a defect in the regulation of IgA1production and glycosylation. IgA1, a predominant subclass of IgA in serum, is characterized by the insertion of two octapeptide repeats in the hinge region between Cα1 and Cα2 domains. This hinge region is absent in IgA2 [90][91]. This hinge region is rich in Ser, Thr, and Pro residues which are potential sites for O-glycosylation with three to six in the amount [90][92][93]. These O-glycans all have an N-acetyl galactosamine core (GalNAc core) attaching to Ser/Thr residuals. These GalNAc cores can exist alone or extend with β 1–3 linked Gal to form disaccharides. These disaccharides can further be sialylated on GalNAc, Gal, or both sugars [94][95]. IgA with abnormal, defective galactosylation of O-glycans is found in patients with IgA nephropathy [96][97]. From IgA-producing cells in peripheral blood suggest that premature sialyation may lead to this abnormal IgA glycosylation [98]. This galactose-deficiency IgA(gd-IgA) can circulate as a monomer or in self-aggregated form. The level of gd-IgA1 in circulation may be partially influenced by exogenous factors, such as bacteria-deprived proteases [99]. It is supposed that the environment, food antigens, or mucosal infections directly or indirectly through TLR signaling trigger the maturation of these B cells. For example, ligation of TLR-9 in B cells with bacterial DNA leads to polyclonal B cell activation, immunoglobulin production, and class switching [100][101]. These signaling increase expression of B cell activation factor (BAFF) and a proliferation-inducing ligand (APRIL) signaling, which co-stimulate B cells at lamina propria maturing into plasma cells [102][103][104][105][106]. These primed B cells, which secret mucosal IgA, are mis-home to circulation where gd-IgA1 immune complexes formed. With circulation, these mis-home B cells can migrate to bone marrow and kidneys and form a tertiary lymphoid organ with focal proliferation [107][108][109].

Figure 1. Pathogenesis of IgA nephropathy with 4 hits theory. Hit 1: synthesis of gd-IgA1; Hit 2: anti-gd-IgA1 antibody production; Hit 3: Circulating IgA immune complex formation; Hit 4: Circulating IgA immune complex deposition to the glomeruli. TLRs: Toll like receptors; BAFF: B-cell activating factor; APRIL: a proliferation-inducing ligand; gd-IgA1: galactose-deficient IgA1; C1GALT1: Core 1 Synthase, Glycoprotein-N-Acetylgalactosamine 3-β-Galactosyltransferase 1; GALNT2: Polypeptide N-Acetylgalactosaminyltransferase 2; ST6GALNAC2: ST6 N-Acetylgalactosaminide α-2,6-Sialyltransferase 2; anti-gd-IgA1 Ab: anti-gd-IgA1 antibody; CNI: calcineurin inhibitor; MMF: mycophenolate mofetil; MPA: mycophenolate acid. Blue dash lines denote undergoing novel treatment in IgA nephropathy.
TLR7 was expressed in abundant these infiltrated CD19+ B cells and is closely related to renal function and histopathological findings. The MyD88 dependent signaling pathway promoted B cell expansion with immunoglobulin secretion and synthesis cytokines (IL-6, IL-12, and IL-1β). IL-6 leads to mesangial proliferation, apoptosis of podocytes, endothelial dysfunction, extracellular matrix production, and renal fibrosis, while IL-12 can recruit and accumulate lymphocytes and induce the secretion of IFN-γ [110].
Disorder of enzyme expression via TLR signaling pathways plays a crucial role in synthesizing gd-IgA1. Overexpression of TLR7 leads to more Polypeptide N-Acetylgalactosaminyltransferase 2(GALNT2) proteins [110]. GALNT2 is the critical determinant of the numbers and pattern of O-glycans to the hinge of IgA1. Glycoprotein-N-acetylgalactosamine 3-β-galactosyltransferase 1(C1GALT1) is responsible for adding galactose to GalNAc via its core 1 β 3-Gal-T-specific molecular chaperone (COSMC). Low C1GALT1/GALNT2 ratio in IgAN with overexpression of GALNT2 leads to higher gd-IgA production. Ligation of TLR-4 in B cells with bacterial PLS can also decrease the activity of C1GALT1 and associated defective galactosylation by methylation of the Cosmc gene [111]. Another similarly reduced expression is activated by TLR9, with a synergy of IL-6 and APRIL to affect the O-glycosyltransferase ST6GALNAC2 [112].
Once the gd-IgA is formed, the second hit is the formation of anti-glycan antibodies. The exposed GalNAc mimics bacterial or viral antigens and can be recognized by specific anti-glycan antibodies. These anti-glycan antibodies lead to the formation of the circulating immune complex [113][114]. Normal IgA1 has a short half-life of about five days and is rapidly catabolized by hepatocytes via sialoglycoprotein receptors (ASGP-R) [115]. However, if the sialic acid is linked to GalNAc or IgA is bound with antibodies, the clearance by hepatocytes may be hindered. Serum gd-IgA1 is bound with antibodies as an immune complex. That is why gd-IgA1 often remains in circulation for a prolonged period. Circulating polymeric complex is known to induce cleavage of the extracellular domain of FcαR (CD89) and form an IgA1-CD89 complex (hit 3). It can further precipitate in mesangial deposition with high affinity [116].
While depositing in the mesangium, these immune complexes stimulate mesangial proliferation and production of IL-6 and TGF-β, which recruit leukocytes with inflammatory reactions and promote glomerular and interstitial fibrosis (hit 4) [117][118][119]. Additionally, activating the complement system involves glomerular inflammation. C3 glomerular staining can be found in over 90–95% of biopsies, while C1q is generally negative. Such dichotomy suggests activating the alternative pathway, likely the lectin pathway [120]. Positive C4 staining with the absence of C1q is found in 40% of biopsies, while mesangial deposition of mannose-binding lectin (MBL) is about 25%. Additionally, L-Ficolin and MBL-associated serine protease are found in positive MBL staining biopsies. These findings suggest the activation of the lectin pathway, and patients having this pattern suffered from more severe histologic damage and more proteinuria. Proteins, including factor B, factor H, and factor H related proteins (FHR), of the alternative pathway, were also found in some biopsies. Accumulating data implied that, contrary to factor H terminating activation, FHR-1 and FHR-5 amplify alternative pathway activation by competing with factor H. Elevation of FHR1 and FHR5 in circulation are correlated with the activity of IgAN, and high FHR5 level is associated with poor response to immunosuppressant [121][122].
Defective antigen handling by mononuclear cells in peripheral blood of patients with IgAN increases the expression of C-X3-C Motif Chemokine Receptor 1 (CX3CR1). Meanwhile, glomerular and urinary fractalkine, the ligand of CX3CR1, in patients with IgAN is also present in a high amount. Therefore, transmigration of these CX3CR1+lymphocytes into the tissue will damage the endothelial cell and role in vascular injury and hematuria [123].
It was reveal, in contrast to the other two primary glomerular diseases, a highly complex polygenic architecture in IgA nephropathy with nearly 20 genome-wide significant loci of minor to moderate effects. Like other major immune-mediated glomerular diseases, the MHC locus on chromosome 6p21 strongly correlates with genetic susceptibility. HLA-DQA1*0101 and HLA-DQB1*0301 appear as risk alleles, while HLA-DQA1*0102 and HLA-DQB1*0201 reveal to be protective [124]. Additionally, the discovery of non-MHC loci reinforced the roles of innate and adaptive immunity in pathogenesis. The HORMAD2-LIF-OSM locus encodes two mucosal immunity and inflammation cytokines, and the IgAN risk allele in this locus has shown a concordant effect on the risk of tonsillitis and tonsillectomy [125][126]. The TNFSF13 locus encodes APRIL involving the regulation of IgA production [127]. DEF locus encodes human antimicrobial peptides called α-defensins 1 and 3 (DEFA1 and DEFA3), and a low copy number increases the risk for IgAN [128]. CARD9 is a proinflammatory molecule promoting the activation of the NF-κB pathway, and this risk variant provides the genetic evidence of the NF-κB pathway in the pathogenesis of IgAN [124]. The discovery of complement system-related loci includes a variant on chromosome 1q32 at the CFH locus and the ITGAM-ITGAX locus [124][125]. Deletion of CHF1 and CHF3, which encode factor H-related peptides, show protection to enhance the effect of factor H in the alternative pathway. ITGAM and ITGAX, fixed in East-Asian populations, encode leukocyte-specific integrin αM and αX of complement receptor 3 and 4 involving the adhesion, migration of leukocytes, and phagocytosis of macrophages. GWAS also reveals quantitative endophenotypes in IgAN. Variants with lower expressions of C1GALT1 encoding core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1, and C1GALT1C1 encoding COSMC lead to increased production of gd-IgA1 [129]. It can provide a powerful tool to detect the earliest molecular participating factors of diseases and facilitate improved disease classification with complex traits based on molecular mechanisms. However, current GWAS results of IgAN are limited to East-Asian and European cohorts and can only explain 7% of the disease risks. More diverse populations and the discovery of more risk variants are expected.
Although the immune system prominently involves the pathogenesis, clinical results from the immunosuppressant treatment are not inspiring. Glucocorticoids in several small trials showed a reduction in proteinuria but no benefit in remission rate. In the Supportive Versus Immunosuppressive Therapy of Progressive IgA nephropathy (STOP) IgAN trial, the treated group received glucocorticoid as monotherapy while GFR ≥ 60 mL/min per 1.73 m2 or glucocorticoids plus cyclophosphamide for three months and azathioprine from month 4 to 36 if GFR = 30–59 mL/min per 1.73 m2. Although with a significant reduction in proteinuria production, there was no effect on the decline of renal functions [130]. A retrospective analysis in Europe reported glucocorticoid treatment significantly reduces proteinuria with better renal survival, even if eGFR ≤ 50 mL/min per 1.73 m2, and the benefit is positively correlated to the level of proteinuria. The prognostic predictivity of the MEST score was blunted after immunotherapy [131]. The Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) assigned patients with proteinuria more than 1 g/day to symptomatic treatment or oral methylprednisolone. The benefit could be seen in the treated group with better primary endpoints (death, ESRD, or decline in renal function). Still, the study was interrupted early owing to excessive infectious events [132]. Small trials with Mycophenolate mofetil and hydroxychloroquine revealed mixed results in Asian groups [133][134]. A meta-analysis of RCTs for treatment with the mycophenolate mofetil in IgA showed no additional benefit or adverse events than other immunosuppressants [135]. Even rituximab, a monoclonal antibody against CD20+ B cells, does not improve proteinuria and renal function [136]. It were interceded with rituximab for invalid against plasma cells which produce the gd-IgA predominantly [137][138]. There are ongoing novel agent trials, including budesonide (non-absorbable steroid, targeting the intestinal mucosal immune system) [139], adrenocorticotropic hormone (synthetic ACTH) [140], Blisibimod (NCT02052219, withdrawn), and atacicept (NCT02808429, targeting BAFF and APRIL signaling pathways), bortezomib (Proteasome, targeting plasma cells) [141], and complement inhibitors (C5a receptor inhibitor Avacopan, NCT02384317; Eculizumab [142], a humanized monoclonal antibody that blocks C5 activation). Although a retrospective cohort analysis in Japan revealed benefits from tonsillectomy for IgAN, with improved renal survival rate, all need more RCTs for validating their therapeutic effects [143].
3. Membranous Nephropathy
Membranous nephropathy (MN) occurs at all ages but predominantly among males, with a mean age of diagnosis at the fifties and sixties. It is the first leading cause, other than diabetes, of nephrotic syndrome in Caucasian adults [144][145]. About four-fifth is idiopathic, and the remaining cases are secondary to medications or other diseases, such as systemic lupus erythematosus, viral hepatitis, or malignancies. About one-third of the patients with pMN undergo spontaneous remission. Nonetheless, it is still the second and third leading cause of end-stage renal disease amid primary glomerular diseases in America and Europe, respectively [146][147].
MN is an autoimmune disease, presenting with immune complex deposition between the podocyte and the glomerular basement membrane. With the damage of integrity of podocytes, a large amount of protein is lost in the urine. Heavy proteinuria brings about hypoalbuminemia, anasarca, and hypercoagulability. With persistent proteinuria, about 40–50% of patients develop renal failure within ten years.
During the 1970s, MN was supposed to be an immune complex associated with nephropathy. Until 2002, the first antigen of MN in humans, neutral endopeptidase (NEP), was discovered from a case of congenital MN [148]. By mass spectrometric analysis of electrophoretic gel band from the serum of patients with MN, M-type phospholipase A2 receptor (PLA2R) was identified. Autoantibodies against PLA2R can be identified in 70–80% of patients with primary MN [149]. PLA2R is a transmembrane glycoprotein, present in large amounts on the apical surface of podocyte processes and may be shed into urine. Its specific function in podocytes is unclear. It is known that group IB secretory phospholipase A2(sPLA2 IB) via PLA2R is toxic and can induce human podocyte apoptosis [150]. Auto-antibodies, without epitope spreading, targeting the N-terminal cysteine-rich (CysR) domain of PLA2R only contribute better prognosis than those with epitope spreading to C-type lectin-like domains (CTLD) [151][152]. Thrombospondin type 1 domain-containing 7A(THSD7A) is another multidomain transmembrane glycoprotein identified by mass spectrometry. THSD7A is found in 2–3% of primary membranous nephropathy (pMN) [153]. By similar model and techniques, other candidates, including EXT1/EXT2 [154][155], NELL1 [156], SEMA3B [157], NCAM1 [158], PCDH7 [159], and HTRA1 [160] were found. Some of them are found concomitantly in the lupus MN or malignancy-associated MN simultaneously, and some remain lack the corresponding antibody. Their roles as real antigens or as biomarkers are still controversial.
It was suggested that dysregulation in immune phenotype, characterized by a decreased Treg number and increased plasma cell/regulatory B cells, is associated with pMN [161]. Predisposing factors to autoantibody formation include genetic susceptibility (for example, variant in HLA-D), or alternation in antigen expression by exogenous factors (e.g., air pollution, infections) [162][163][164][165]. The most extensive multi-ethnic genome-wide association study (GWAS) in MN found significant loci with genetic effects encoding two transcriptional master regulators of inflammation (for example, NFKB1 and IRF4). These findings underscore the effects of transcriptional regulation in an inflammatory response and the implication of the role of infection in disease induction [165]. In China, it was found that long-term exposure to air pollution increases the incidence of MN [166]. Like NEP as a trigger to induce allo-autoantibody, associated MN has been found in a case of Pompe disease who received enzyme replacement therapy and developed an alloimmune response to the recombinant human arylsulfatase B (rhASB) [167].
The framework of injury to podocytes with proteinuria production unveiled the prelude by precipitation of subepithelial immune deposits, followed by activation of the complement system and assembly of the terminal complement component C5b-9. IgG and C3 are present in disease-established situations, but C1q is usually weak or absent, as observed by immunofluorescence staining, implying the minor role of the classical complement pathway [168]. As the disease progress, the IgG subclass switch from IgG1 and IgG3 at the initial stage to enriched IgG4 at the last stage, activation of the complement system may also have a similar switch from the classical to the alternative or lectin pathway. Besides that the complement membrane attacks complex C5b-9, the upregulation of C3a and C5a receptors (C3aR1 and C5aR1) is shown on podocytes of pMN. There is a positive correlation between urinary C5a and the level of anti-PLA2 antibodies or proteinuria [169][170][171][172]. Activation of C3aR1 and C5aR1 alone with MAC insertion, mediated by aspartic protease and cysteine protease, resulting in proteolysis of synaptopodin, NEPH1, and dynamin (Figure 2) [173].
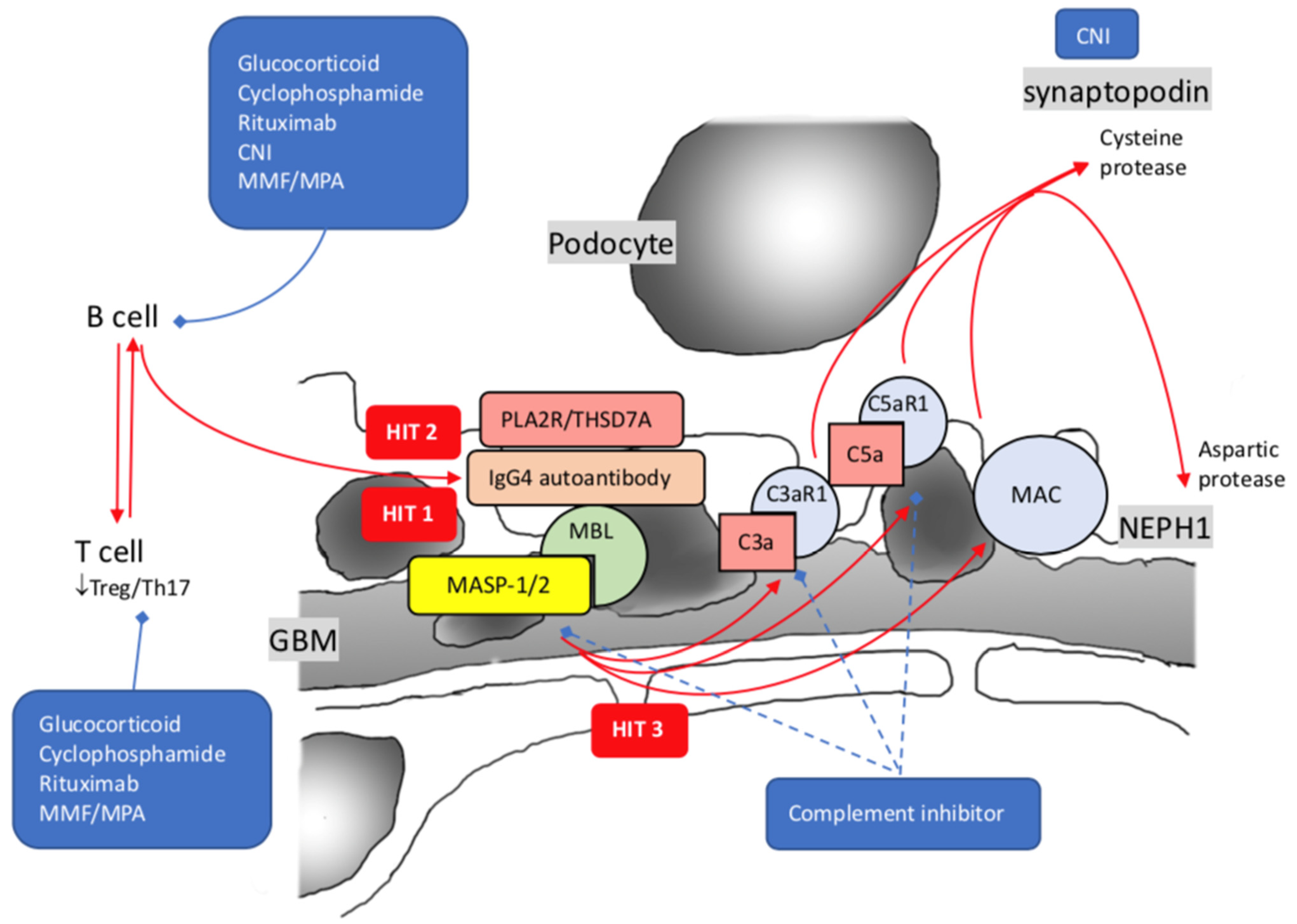
Figure 2. Pathogenesis of idiopathic membranous nephropathy with 3 hits theory. Hit 1: Synthesis of autoantibodies against PLA2R/THSD7A; Hit 2: IgG4 predominant autoimmunization against PLA2R/THSD7A by class shift; Hit 3: Increased expression of C3a1 and C5a1 alone with MAC. PLA2R: Phospholipase-A2-Receptor; THSD7A: Thrombospondin type I domain-containing 7A; MBL: mannose-binding lectin; MASP-1/2: mannose-associated serine protease 1 and mannose-associated serine protease 2; MAC: membrane attack complex; CNI: calcineurin inhibitor; MMF: mycophenolate mofetil; MPA: mycophenolate acid; blue dash lines imply possible novel treatments.
References
- Eddy, A.A.; Symons, J.M. Nephrotic syndrome in childhood. Lancet 2003, 362, 629–639.
- Floege, J.; Amann, K. Primary glomerulonephritides. Lancet 2016, 387, 2036–2048.
- Mathieson, P.W. Minimal change nephropathy and focal segmental glomerulosclerosis. Semin. Immunopathol. 2007, 29, 415–426.
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68.
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411.
- Maas, R.J.; Deegens, J.K.; Smeets, B.; Moeller, M.J.; Wetzels, J.F. Minimal change disease and idiopathic FSGS: Manifestations of the same disease. Nat. Rev. Nephrol. 2016, 12, 768–776.
- Deegens, J.K.; Dijkman, H.B.; Borm, G.F.; Steenbergen, E.J.; van den Berg, J.G.; Weening, J.J.; Wetzels, J.F. Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int. 2008, 74, 1568–1576.
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214.
- Tejani, A. Morphological transition in minimal change nephrotic syndrome. Nephron 1985, 39, 157–159.
- Schwimmer, J.A.; Markowitz, G.S.; Valeri, A.; Appel, G.B. Collapsing glomerulopathy. Semin. Nephrol. 2003, 23, 209–218.
- Shalhoub, R.J. Pathogenesis of lipoid nephrosis: A disorder of T-cell function. Lancet 1974, 2, 556–560.
- MacDonald, N.E.; Wolfish, N.; McLaine, P.; Phipps, P.; Rossier, E. Role of respiratory viruses in exacerbations of primary nephrotic syndrome. J. Pediatr. 1986, 108, 378–382.
- Kim, S.R.; Lee, S.B.; Kim, I.Y.; Lee, D.W.; Rhee, H.; Seong, E.Y.; Song, S.H.; Kwak, I.S. Relapse of minimal change disease following infection with the 2009 pandemic influenza (H1N1) virus. Clin. Exp. Nephrol. 2012, 16, 329–332.
- Lin, C.Y.; Hsu, H.C. Histopathological and immunological studies in spontaneous remission of nephrotic syndrome after intercurrent measles infection. Nephron 1986, 42, 110–115.
- Aggarwal, N.; Batwara, R.; McCarthy, E.T.; Sharma, R.; Sharma, M.; Savin, V.J. Serum permeability activity in steroid-resistant minimal change nephrotic syndrome is abolished by treatment of Hodgkin disease. Am. J. Kidney Dis. 2007, 50, 826–829.
- Peces, R.; Sánchez, L.; Gorostidi, M.; Alvarez, J. Minimal change nephrotic syndrome associated with Hodgkin’s lymphoma. Nephrol. Dial. Transplant. 1991, 6, 155–158.
- Rizvi, S.N.; Vaishnava, H. Cyclophosphamide in minimal-change nephrotic syndrome. Lancet 1972, 2, 537–538.
- Black, D.A.; Rose, G.; Brewer, D.B. Controlled trial of prednisone in adult patients with the nephrotic syndrome. Br. Med. J. 1970, 3, 421–426.
- Shimada, M.; Araya, C.; Rivard, C.; Ishimoto, T.; Johnson, R.J.; Garin, E.H. Minimal change disease: A “two-hit” podocyte immune disorder? Pediatr. Nephrol. 2011, 26, 645–649.
- Wakem, P.; Burns, R.P.; Ramirez, F.; Zlotnick, D.; Ferbel, B.; Haidaris, C.G.; Gaspari, A.A. Allergens and irritants transcriptionally upregulate CD80 gene expression in human keratinocytes. J. Investig. Dermatol. 2000, 114, 1085–1092.
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397.
- Khullar, B.; Balyan, R.; Oswal, N.; Jain, N.; Sharma, A.; Abdin, M.Z.; Bagga, A.; Bhatnagar, S.; Wadhwa, N.; Natchu, U.C.M.; et al. Interaction of CD80 with Neph1: A potential mechanism of podocyte injury. Clin. Exp. Nephrol. 2018, 22, 508–516.
- Shimada, M.; Ishimoto, T.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Wymer, D.T.; Yamabe, H.; Mathieson, P.W.; Saleem, M.A.; et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappaB-dependent pathway. Nephrol. Dial. Transplant. 2012, 27, 81–89.
- Ishimoto, T.; Shimada, M.; Gabriela, G.; Kosugi, T.; Sato, W.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.; Maruyama, S.; Garin, E.H.; et al. Toll-like receptor 3 ligand, polyIC, induces proteinuria and glomerular CD80, and increases urinary CD80 in mice. Nephrol. Dial. Transplant. 2013, 28, 1439–1446.
- Li, S.; Liu, Y.; He, Y.; Rong, W.; Zhang, M.; Li, L.; Liu, Z.; Zen, K. Podocytes present antigen to activate specific T cell immune responses in inflammatory renal disease. J. Pathol. 2020, 252, 165–177.
- Tsuji, S.; Kimata, T.; Yamanouchi, S.; Kitao, T.; Kino, J.; Suruda, C.; Kaneko, K. Regulatory T cells and CTLA-4 in idiopathic nephrotic syndrome. Pediatr. Int. 2017, 59, 643–646.
- Kimata, T.; Tsuji, S.; Kino, J.; Kitao, T.; Yamanouchi, S.; Kaneko, K. Close association between proteinuria and regulatory T cells in patients with idiopathic nephrotic syndrome. Pediatr. Nephrol. 2013, 28, 667–669.
- Cara-Fuentes, G.; Wasserfall, C.H.; Wang, H.; Johnson, R.J.; Garin, E.H. Minimal change disease: A dysregulation of the podocyte CD80-CTLA-4 axis? Pediatr. Nephrol. 2014, 29, 2333–2340.
- Matsumoto, K. Decreased release of IL-10 by monocytes from patients with lipoid nephrosis. Clin. Exp. Immunol. 1995, 102, 603–607.
- Chen, W.; Wu, Y.; Zhang, G.; Wang, M.; Yang, H.; Li, Q. Gammadeltat Cells Exacerbate Podocyte Injury via the CD28/B7-1-Phosphor-SRC Kinase Pathway. Biomed. Res. Int. 2018, 2018, 5647120.
- Zhang, S.-Y.; Kamal, M.; Dahan, K.; Pawlak, A.; Ory, V.; Desvaux, D.; Audard, V.; Candelier, M.; BenMohamed, F.; Matignon, M.; et al. c-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci. Signal 2010, 3, ra39.
- Liu, Y.; Su, L.; Lin, Q.; Han, Y.; You, P.; Fan, Q. Induction of C-Mip by IL-17 Plays an Important Role in Adriamycin-Induced Podocyte Damage. Cell Physiol. Biochem. 2015, 36, 1274–1290.
- May, C.J.; Welsh, G.I.; Chesor, M.; Lait, P.J.; Schewitz-Bowers, L.P.; Lee, R.W.J.; Saleem, M.A. Human Th17 cells produce a soluble mediator that increases podocyte motility via signaling pathways that mimic PAR-1 activation. Am. J. Physiol. Renal. Physiol. 2019, 317, F913–F921.
- Liu, L.L.; Qin, Y.; Cai, J.F.; Wang, H.Y.; Tao, J.L.; Li, H.; Chen, L.M.; Li, M.X.; Li, X.M.; Li, X.W. Th17/Treg imbalance in adult patients with minimal change nephrotic syndrome. Clin. Immunol. 2011, 139, 314–320.
- Stachowski, J.; Barth, C.; Michałkiewicz, J.; Krynicki, T.; Jarmoliński, T.; Runowski, D.; Lewandowska-Stachowiak, M.; Zaniew, M.; Warzywoda, A.; Bortkiewicz, E.; et al. Th1/Th2 balance and CD45-positive T cell subsets in primary nephrotic syndrome. Pediatr. Nephrol. 2000, 14, 779–785.
- Webendörfer, M.; Reinhard, L.; Stahl, R.A.K.; Wiech, T.; Mittrücker, H.-W.; Harendza, S.; Hoxha, E. Rituximab Induces Complete Remission of Proteinuria in a Patient With Minimal Change Disease and No Detectable B Cells. Front. Immunol. 2020, 11, 586012.
- Fenoglio, R.; Sciascia, S.; Beltrame, G.; Mesiano, P.; Ferro, M.; Quattrocchio, G.; Menegatti, E.; Roccatello, D. Rituximab as a front-line therapy for adult-onset minimal change disease with nephrotic syndrome. Oncotarget 2018, 9, 28799–28804.
- Takemura, S.; Klimiuk, P.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T cell activation in rheumatoid synovium is B cell dependent. J. Immunol. 2001, 167, 4710–4718.
- Silverman, G.J.; Weisman, S. Rituximab therapy and autoimmune disorders: Prospects for anti-B cell therapy. Arthritis Rheum. 2003, 48, 1484–1492.
- Lipsky, P.E. Systemic lupus erythematosus: An autoimmune disease of B cell hyperactivity. Nat. Immunol. 2001, 2, 764–766.
- Silverman, G.J. Anti-CD20 therapy and autoimmune disease: Therapeutic opportunities and evolving insights. Front. Biosci. 2007, 12, 2194–2206.
- MacAulay, A.E.; DeKruyff, R.H.; Goodnow, C.; Umetsu, D.T. Antigen-specific B cells preferentially induce CD4+ T cells to produce IL-4. J. Immunol. 1997, 158, 4171–4179.
- Clark, R.; Kupper, T. Old meets new: The interaction between innate and adaptive immunity. J. Investig. Dermatol. 2005, 125, 629–637.
- Sinha, A.; Bagga, A. Rituximab therapy in nephrotic syndrome: Implications for patients’ management. Nat. Rev. Nephrol. 2013, 9, 154–169.
- Hansrivijit, P.; Cheungpasitporn, W.; Thongprayoon, C.; Ghahramani, N. Rituximab therapy for focal segmental glomerulosclerosis and minimal change disease in adults: A systematic review and meta-analysis. BMC Nephrol. 2020, 21, 134.
- Brown, L.C.; Jobson, M.A.; Schober, F.P.; Chang, E.H.; Falk, R.J.; Nachman, P.H.; Pendergraft, W.F. The Evolving Role of Rituximab in Adult Minimal Change Glomerulopathy. Am. J. Nephrol. 2017, 45, 365–372.
- Bai, A.; Robson, S. Beyond ecto-nucleotidase: CD39 defines human Th17 cells with CD161. Purinergic Signal. 2015, 11, 317–319.
- Ravani, P.; Bonanni, A.; Rossi, R.; Caridi, G.; Ghiggeri, G.M. Anti-CD20 Antibodies for Idiopathic Nephrotic Syndrome in Children. Clin. J. Am. Soc. Nephrol. 2016, 11, 710–720.
- van de Veerdonk, F.L.; Lauwerys, B.; Marijnissen, R.J.; Timmermans, K.; Di Padova, F.; Koenders, M.I.; Gutierrez-Roelens, I.; Durez, P.; Netea, M.G.; van der Meer, J.W.M.; et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011, 63, 1507–1516.
- Orikasa, M.; Matsui, K.; Oite, T.; Shimizu, F. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J. Immunol. 1988, 141, 807–814.
- Topham, P.S.; Kawachi, H.; Haydar, S.A.; Chugh, S.; Addona, T.A.; Charron, K.B.; Holzman, L.B.; Shia, M.; Shimizu, F.; Salant, D.J. Nephritogenic mAb 5-1-6 is directed at the extracellular domain of rat nephrin. J. Clin. Investig. 1999, 104, 1559–1566.
- Takeuchi, K.; Naito, S.; Kawashima, N.; Ishigaki, N.; Sano, T.; Kamata, K.; Takeuchi, Y. New Anti-Nephrin Antibody Mediated Podocyte Injury Model. Using a C57BL/6 Mouse Strain. Nephron 2018, 138, 71–87.
- Coward, R.J.; Foster, R.R.; Patton, D.; Ni, L.; Lennon, R.; Bates, D.O.; Harper, S.J.; Mathieson, P.W.; Saleem, M.A. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J. Am. Soc. Nephrol. 2005, 16, 629–637.
- Wernerson, A. Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol. Dial. Transplant. 2003, 18, 70–76.
- Watts, A.J.; Keller, K.H.; Lerner, G.; Rosales, I.; Collins, A.B.; Sekulic, M.; Waikar, S.S.; Chandraker, A.; Riella, L.V.; Alexander, M.P.; et al. Discovery of Autoantibodies Targeting Nephrin in Minimal Change Disease Supports a Novel Autoimmune Etiology. J. Am. Soc. Nephrol. 2022, 33, 238–252.
- Kienzl-Wagner, K.; Waldegger, S.; Schneeberger, S. Disease Recurrence-The Sword of Damocles in Kidney Transplantation for Primary Focal Segmental Glomerulosclerosis. Front. Immunol. 2019, 10, 1669.
- McCarthy, E.T.; Sharma, M.; Savin, V.J. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2115–2121.
- Chebotareva, N.; Vinogradov, A.; Cao, V.; Gindis, A.; Berns, A.; Alentov, I.; Sergeeva, N. Serum levels of plasminogen activator urokinase receptor and cardiotrophin-like cytokine factor 1 in patients with nephrotic syndrome. Clin. Nephrol. 2022, 97, 103–110.
- Doublier, S.; Zennaro, C.; Musante, L.; Spatola, T.; Candiano, G.; Bruschi, M.; Besso, L.; Cedrino, M.; Carraro, M.; Ghiggeri, G.M.; et al. Soluble CD40 ligand directly alters glomerular permeability and may act as a circulating permeability factor in FSGS. PLoS ONE 2017, 12, e0188045.
- Li, Y.; Kang, Y.S.; Dai, C.; Kiss, L.P.; Wen, X.; Liu, Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am. J. Pathol. 2008, 172, s299–s308.
- Huber, T.B.; Benzing, T. The slit diaphragm: A signaling platform to regulate podocyte function. Curr. Opin. Nephrol. Hypertens. 2005, 14, 211–216.
- Verma, R.; Wharram, B.; Kovari, I.; Kunkel, R.; Nihalani, D.; Wary, K.; Wiggins, R.; Killen, P.; Holzman, L.B. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J. Biol. Chem. 2003, 278, 20716–20723.
- Lahdenperä, J.; Kilpeläinen, P.; Liu, X.L.; Pikkarainen, T.; Reponen, P.; Ruotsalainen, V.; Tryggvason, K. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 2003, 64, 404–413.
- Li, H.; Lemay, S.; Aoudjit, L.; Kawachi, H.; Takano, T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 2004, 15, 3006–3015.
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566.
- Blasutig, I.M.; New, L.A.; Thanabalasuriar, A.; Dayarathna, T.K.; Goudreault, M.; Quaggin, S.E.; Li, S.S.; Gruenheid, S.; Jones, N.; Pawson, T. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol. Cell Biol. 2008, 28, 2035–2046.
- Uchida, K.; Suzuki, K.; Iwamoto, M.; Kawachi, H.; Ohno, M.; Horita, S.; Nitta, K. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int. 2008, 73, 926–932.
- Ohashi, T.; Uchida, K.; Asamiya, Y.; Tsuruta, Y.; Ohno, M.; Horita, S.; Nitta, K. Phosphorylation status of nephrin in human membranous nephropathy. Clin. Exp. Nephrol. 2010, 14, 51–55.
- New, L.A.; Martin, C.E.; Scott, R.; Platt, M.J.; Chahi, A.K.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435.
- Wagner, M.C.; Rhodes, G.; Wang, E.; Pruthi, V.; Arif, E.; Saleem, M.A.; Wean, S.E.; Garg, P.; Verma, R.; Holzman, L.B.; et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 2008, 283, 35579–35589.
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Hattori, S. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 2008, 283, 9177–9186.
- Arif, E.; Rathore, Y.S.; Kumari, B.; Ashish, F.; Wong, H.N.; Holzman, L.B.; Nihalani, D. Slit diaphragm protein Neph1 and its signaling: A novel therapeutic target for protection of podocytes against glomerular injury. J. Biol. Chem. 2014, 289, 9502–9518.
- Fukusumi, Y.; Zhang, Y.; Yamagishi, R.; Oda, K.; Watanabe, T.; Matsui, K.; Kawachi, H. Nephrin-Binding Ephrin-B1 at the Slit Diaphragm Controls Podocyte Function through the JNK Pathway. J. Am. Soc. Nephrol. 2018, 29, 1462–1474.
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Choi, H.Y.; Chang, J.H.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427.
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; A Correia, L.; Tong, H.-Q.; Mathis, B.J.; Rodríguez-Pérez, J.-C.; Allen, P.G.; Beggs, A.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256.
- Buvall, L.; Wallentin, H.; Sieber, J.; Andreeva, S.; Choi, H.Y.; Mundel, P.; Greka, A. Synaptopodin Is a Coincidence Detector of Tyrosine versus Serine/Threonine Phosphorylation for the Modulation of Rho Protein Crosstalk in Podocytes. J. Am. Soc. Nephrol. 2017, 28, 837–851.
- Johansen, K.L.; Chertow, G.M.; Foley, R.N.; Gilbertson, D.T.; Herzog, C.A.; Ishani, A.; Israni, A.K.; Ku, E.; Kurella Tamura, M.; Li, S.; et al. 2020 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 2021, 77 (Suppl. 1), A7–A8.
- Brown, E.J.; Pollak, M.R.; Barua, M. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int. 2014, 85, 1030–1038.
- Lepori, N.; Zand, L.; Sethi, S.; Fernandez-Juarez, G.; Fervenza, F.C. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin. Kidney J. 2018, 11, 179–190.
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289.
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517.
- Friedman, D.J.; Pollak, M.R. APOL1 Nephropathy: From Genetics to Clinical Applications. Clin. J. Am. Soc. Nephrol. 2021, 16, 294–303.
- Nourbakhsh, N.; Mak, R.H. Steroid-resistant nephrotic syndrome: Past and current perspectives. Pediatric. Health Med. Ther. 2017, 8, 29–37.
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779.
- Ding, W.Y.; Koziell, A.; McCarthy, H.J.; Bierzynska, A.; Bhagavatula, M.K.; Dudley, J.A.; Inward, C.D.; Coward, R.; Tizard, J.; Reid, C.; et al. Initial steroid sensitivity in children with steroid-resistant nephrotic syndrome predicts post-transplant recurrence. J. Am. Soc. Nephrol. 2014, 25, 1342–1348.
- Julian, B.A.; Woodford, S.Y.; Baehler, R.W.; McMorrow, R.G.; Wyatt, R.J. Familial clustering and immunogenetic aspects of IgA nephropathy. Am. J. Kidney Dis. 1988, 12, 366–370.
- Sato, Y.; Tsukaguchi, H.; Higasa, K.; Kawata, N.; Inui, K.; Linh, T.N.T.; Quynh, T.T.H.; Yoshihiko, I.; Koiwa, F.; Yoshimura, A. Positive renal familial history in IgA nephropathy is associated with worse renal outcomes: A single-center longitudinal study. BMC Nephrol. 2021, 22, 230.
- Rodrigues, J.C.; Haas, M.; Reich, H.N. IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 677–686.
- Magistroni, R.; D’Agati, V.D.; Appel, G.B.; Kiryluk, K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015, 88, 974–989.
- Baenziger, J.; Kornfeld, S. Structure of the carbohydrate units of IgA1 immunoglobulin. II. Structure of the O-glycosidically linked oligosaccharide units. J. Biol. Chem. 1974, 249, 7270–7281.
- Tomana, M.; Niedermeier, W.; Mestecky, J.; Skvaril, F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry 1976, 13, 325–328.
- Tarelli, E.; Smith, A.C.; Hendry, B.M.; Challacombe, S.; Pouria, S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr. Res. 2004, 339, 2329–2335.
- Franc, V.; Řehulka, P.; Raus, M.; Stulík, J.; Novak, J.; Renfrow, M.B.; Šebela, M. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: Role of cysteine alkylation during sample processing. J. Proteom. 2013, 92, 299–312.
- Field, M.C.; Dwek, R.A.; Edge, C.J.; Rademacher, T.W. O-linked oligosaccharides from human serum immunoglobulin A1. Biochem. Soc. Trans. 1989, 17, 1034–1035.
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcalpha receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272.
- Ohyama, Y.; Renfrow, M.; Novak, J.; Takahashi, K. Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know. J. Clin. Med. 2021, 10, 3467.
- Allen, A.C.; Bailey, E.M.; Brenchley, P.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973.
- Takahashi, K.; Raska, M.; Horynová, M.S.; Hall, S.D.; Poulsen, K.; Kilian, M.; Hiki, Y.; Yuzawa, Y.; Moldoveanu, Z.; Julian, B.A.; et al. Enzymatic sialylation of IgA1 O-glycans: Implications for studies of IgA nephropathy. PLoS ONE 2014, 9, e99026.
- Wang, L.; Wang, H.; Li, X.; Fan, J. Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA Nephropathy. Sci. Rep. 2016, 6, 30964.
- Blaas, S.H.; Stieber-Gunckel, M.; Falk, W.; Obermeier, F.; Rogler, G. CpG-oligodeoxynucleotides stimulate immunoglobulin A secretion in intestinal mucosal B cells. Clin. Exp. Immunol. 2009, 155, 534–540.
- Cognasse, F.; Acquart, S.; Béniguel, L.; Sabido, O.; Chavarin, P.; Genin, C.; Garraud, O. Differential production of immunoglobulin classes and subclasses by mucosal-type human B-lymphocytes exposed in vitro to CpG oligodeoxynucleotides. Clin. Chem. Lab. Med. 2005, 43, 22–31.
- Takahara, M.; Nagato, T.; Nozaki, Y.; Kumai, T.; Katada, A.; Hayashi, T.; Harabuchi, Y. A proliferation-inducing ligand (APRIL) induced hyper-production of IgA from tonsillar mononuclear cells in patients with IgA nephropathy. Cell Immunol. 2019, 341, 103925.
- Martín-Penagos, L.; Benito-Hernández, A.; Segundo, D.S.; Sango, C.; Azueta, A.; Gómez-Román, J.; Fernández-Fresnedo, G.; López-Hoyos, M.; Ruiz, J.C.; Rodrigo, E. A proliferation-inducing ligand increase precedes IgA nephropathy recurrence in kidney transplant recipients. Clin. Transplant. 2019, 33, e13502.
- Xin, G.; Shi, W.; Xu, L.-X.; Su, Y.; Yan, L.-J.; Li, K.-S. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J. Nephrol. 2013, 26, 683–690.
- Im, J.; Baik, J.E.; Lee, D.; Park, O.J.; Park, D.H.; Yun, C.H.; Han, S.H. Bacterial Lipoproteins Induce BAFF Production via TLR2/MyD88/JNK Signaling Pathways in Dendritic Cells. Front. Immunol. 2020, 11, 564699.
- Hardenberg, G.; Planelles, L.; Schwarte, C.M.; van Bostelen, L.; Le Huong, T.; Hahne, M.; Medema, J.P. Specific TLR ligands regulate APRIL secretion by dendritic cells in a PKR-dependent manner. Eur. J. Immunol. 2007, 37, 2900–2911.
- Batra, A.; Smith, A.C.; Feehally, J.; Barratt, J. T-cell homing receptor expression in IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 2540–2548.
- March, A.K.-D.; Bene, M.C.; Renoult, E.; Kessler, M.; Faure, G.C.; Kolopp-Sarda, M.N. Enhanced expression of L-selectin on peripheral blood lymphocytes from patients with IgA nephropathy. Clin. Exp. Immunol. 1999, 115, 542–546.
- Buren, M.; Yamashita, M.; Suzuki, Y.; Tomino, Y.; Emancipator, S.N. Altered expression of lymphocyte homing chemokines in the pathogenesis of IgA nephropathy. Contrib. Nephrol. 2007, 157, 50–55.
- Zheng, N.; Xie, K.; Ye, H.; Dong, Y.; Wang, B.; Luo, N.; Fan, J.; Tan, J.; Chen, W.; Yu, X. TLR7 in B cells promotes renal inflammation and Gd-IgA1 synthesis in IgA nephropathy. JCI Insight 2020, 5, e136965.
- Qin, W.; Zhong, X.; Fan, J.M.; Zhang, Y.J.; Liu, X.R.; Ma, X.Y. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1608–1614.
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 Stimulation Induces Aberrant Expression of a Proliferation-Inducing Ligand by Tonsillar Germinal Center B Cells in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238.
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677.
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81.
- Roggenbuck, D.; Mytilinaiou, M.G.; Lapin, S.V.; Reinhold, D.; Conrad, K. Asialoglycoprotein receptor (ASGPR): A peculiar target of liver-specific autoimmunity. Autoimmun. Highlights 2012, 3, 119–125.
- Launay, P.; Grossetête, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009.
- Ebefors, K.; Liu, P.; Lassén, E.; Elvin, J.; Candemark, E.; Levan, K.; Haraldsson, B.; Nyström, J. Mesangial cells from patients with IgA nephropathy have increased susceptibility to galactose-deficient IgA1. BMC Nephrol. 2016, 17, 40.
- Zhao, Y.-F.; Zhu, L.; Liu, L.-J.; Shi, S.-F.; Lv, J.-C.; Zhang, H. Pathogenic role of glycan-specific IgG antibodies in IgA nephropathy. BMC Nephrol. 2017, 18, 301.
- Wu, M.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, P.-W.; Chen, Y.-L.; Chiu, H.-C.; Liu, K.-H.; Lee, W.-C.; Li, C.-J. The Emerging Role of Pathogenesis of IgA Nephropathy. J. Clin. Med. 2018, 7, 225.
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; Van Gijlswijk-Janssen, D.J.; Stahl, G.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734.
- Medjeral-Thomas, N.R.; Lomax-Browne, H.J.; Beckwith, H.; Willicombe, M.; McLean, A.G.; Brookes, P.; Pusey, C.D.; Falchi, M.; Cook, H.T.; Pickering, M.C. Circulating complement factor H-related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017, 92, 942–952.
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy Is Associated With Low Circulating Mannan-Binding Lectin-Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H-Related Protein-5 (FHR5) Deposition. Kidney Int. Rep. 2018, 3, 426–438.
- Cox, S.N.; Sallustio, F.; Serino, G.; Loverre, A.; Pesce, F.; Gigante, M.; Zaza, G.; Stifanelli, P.F.; Ancona, N.; Schena, F.P. Activated innate immunity and the involvement of CX3CR1-fractalkine in promoting hematuria in patients with IgA nephropathy. Kidney Int. 2012, 82, 548–560.
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196.
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; A Julian, B.; Wyatt, R.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327.
- Feenstra, B.; Bager, P.; Liu, X.; Hjalgrim, H.; A Nohr, E.; Hougaard, D.M.; Geller, F.; Melbye, M. Genome-wide association study identifies variants in HORMAD2 associated with tonsillectomy. J. Med. Genet. 2017, 54, 358–364.
- Yu, X.-Q.; Li, M.; Zhang, H.; Low, H.-Q.; Wei, X.; Wang, J.-Q.; Sun, L.-D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182.
- Ai, Z.; Li, M.; Liu, W.; Foo, J.-N.; Mansouri, O.; Yin, P.; Zhou, Q.; Tang, X.; Dong, X.; Feng, S.; et al. Low alpha-defensin gene copy number increases the risk for IgA nephropathy and renal dysfunction. Sci. Transl. Med. 2016, 8, 345ra88.
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017, 13, e1006609.
- Rauen, T.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Effects of Two Immunosuppressive Treatment Protocols for IgA Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 317–325.
- Tesar, V.; Troyanov, S.; Bellur, S.; Verhave, J.C.; Cook, H.T.; Feehally, J.; Roberts, I.S.; Cattran, D.; Coppo, R.; On behalf of the VALIGA study of the ERA-EDTA Immunonephrology Working Group. Corticosteroids in IgA Nephropathy: A Retrospective Analysis from the VALIGA Study. J. Am. Soc. Nephrol. 2015, 26, 2248–2258.
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442.
- Yang, Y.-Z.; Chen, P.; Liu, L.-J.; Cai, Q.-Q.; Shi, S.-F.; Chen, Y.-Q.; Lv, J.-C.; Zhang, H. Comparison of the effects of hydroxychloroquine and corticosteroid treatment on proteinuria in IgA nephropathy: A case-control study. BMC Nephrol. 2019, 20, 297.
- Maes, B.D.; Oyen, R.; Claes, K.; Evenepoel, P.; Kuypers, D.R.; Vanwalleghem, J.; Van Damme, B.; Vanrenterghem, Y.F.C. Mycophenolate mofetil in IgA nephropathy: Results of a 3-year prospective placebo-controlled randomized study. Kidney Int. 2004, 65, 1842–1849.
- Zheng, J.; Bi, T.; Zhu, L.; Liu, L. Efficacy and safety of mycophenolate mofetil for IgA nephropathy: An updated meta-analysis of randomized controlled trials. Exp. Ther. Med. 2018, 16, 1882–1890.
- Lafayette, R.A.; Canetta, P.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J. Am. Soc. Nephrol. 2017, 28, 1306–1313.
- McHeyzer-Williams, M.G.; Ahmed, R. B cell memory and the long-lived plasma cell. Curr. Opin. Immunol. 1999, 11, 172–179.
- O’Connor, B.P.; Cascalho, M.; Noelle, R.J. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 2002, 195, 737–745.
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127.
- Zand, L.; Canetta, P.; Lafayette, R.; Aslam, N.; Jan, N.; Sethi, S.; Fervenza, F.C. An Open-Label Pilot Study of Adrenocorticotrophic Hormone in the Treatment of IgA Nephropathy at High Risk of Progression. Kidney Int. Rep. 2020, 5, 58–65.
- Hartono, C.; Chung, M.; Perlman, A.S.; Chevalier, J.M.; Serur, D.; Seshan, S.V.; Muthukumar, T. Bortezomib for Reduction of Proteinuria in IgA Nephropathy. Kidney Int. Rep. 2018, 3, 861–866.
- Rosenblad, T.; Rebetz, J.; Johansson, M.; Békássy, Z.; Sartz, L.; Karpman, D. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr. Nephrol. 2014, 29, 2225–2228.
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association Between Tonsillectomy and Outcomes in Patients With Immunoglobulin A Nephropathy. JAMA Netw. Open 2019, 2, e194772.
- Politano, S.A.; Colbert, G.B.; Hamiduzzaman, N. Nephrotic Syndrome. Prim. Care 2020, 47, 597–613.
- Keri, K.C.; Blumenthal, S.; Kulkarni, V.; Beck, L.; Chongkrairatanakul, T. Primary membranous nephropathy: Comprehensive review and historical perspective. Postgrad. Med. J. 2019, 95, 23–31.
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997.
- Cattran, D.C.; Brenchley, P.E. Membranous nephropathy: Integrating basic science into improved clinical management. Kidney Int. 2017, 91, 566–574.
- Debiec, H.; Guigonis, V.; Mougenot, B.; Decobert, F.; Haymann, J.-P.; Bensman, A.; Deschenes, G.; Ronco, P.M. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N. Engl. J. Med. 2002, 346, 2053–2060.
- Beck, L.; Bonegio, R.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 2009, 361, 11–21.
- Pan, Y.; Wan, J.; Liu, Y.; Yang, Q.; Liang, W.; Singhal, P.C.; Saleem, M.A.; Ding, G. sPLA2 IB induces human podocyte apoptosis via the M-type phospholipase A2 receptor. Sci. Rep. 2014, 4, 6660.
- Salant, D.J. Does Epitope Spreading Influence Responsiveness to Rituximab in PLA2R-Associated Membranous Nephropathy? Clin. J. Am. Soc. Nephrol. 2019, 14, 1122–1124.
- Seitz-Polski, B.; Dolla, G.; Payre, C.; Girard, C.; Polidori, J.; Zorzi, K.; Birgy-Barelli, E.; Jullien, P.; Courivaud, C.; Krummel, T.; et al. Epitope Spreading of Autoantibody Response to PLA2R Associates with Poor Prognosis in Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 1517–1533.
- Tomas, N.M.; Beck, L.H.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.-S.; et al. Thrombospondin Type-1 Domain-Containing 7A in Idiopathic Membranous Nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287.
- Ravindran, A.; Moura, M.C.; Fervenza, F.C.; Nasr, S.H.; Alexander, M.P.; Fidler, M.E.; Hernandez, L.P.H.; Zhang, P.; Grande, J.P.; Cornell, L.D.; et al. In Patients with Membranous Lupus Nephritis, Exostosin-Positivity and Exostosin-Negativity Represent Two Different Phenotypes. J. Am. Soc. Nephrol. 2021, 32, 695–706.
- Saïdi, M.; Brochériou, I.; Estève, E.; Tuffet, S.; Amoura, Z.; Miyara, M.; Belenfant, X.; Ulinski, T.; Rouvier, P.; Debiec, H.; et al. The Exostosin Immunohistochemical Status Differentiates Lupus Membranous Nephropathy Subsets with Different Outcomes. Kidney Int. Rep. 2021, 6, 1977–1980.
- Caza, T.N.; Hassen, S.I.; Dvanajscak, Z.; Kuperman, M.; Edmondson, R.; Herzog, C.; Storey, A.; Arthur, J.; Cossey, L.N.; Sharma, S.G.; et al. NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int. 2020, 99, 967–976.
- Sethi, S.; Debiec, H.; Madden, B.; Vivarelli, M.; Charlesworth, M.C.; Ravindran, A.; Gross, L.; Ulinski, T.; Buob, D.; Tran, C.L.; et al. Semaphorin 3B–associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int. 2020, 98, 1253–1264.
- Caza, T.N.; Hassen, S.I.; Kuperman, M.; Sharma, S.G.; Dvanajscak, Z.; Arthur, J.; Edmondson, R.; Storey, A.; Herzog, C.; Kenan, D.J.; et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int. 2020, 100, 171–181.
- Sethi, S.; Madden, B.; Debiec, H.; Morelle, J.; Charlesworth, M.C.; Gross, L.; Negron, V.; Buob, D.; Chaudhry, S.; Jadoul, M.; et al. Protocadherin 7–Associated Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1249–1261.
- Al-Rabadi, L.F.; Caza, T.; Trivin-Avillach, C.; Rodan, A.R.; Andeen, N.; Hayashi, N.; Williams, B.; Revelo, M.P.; Clayton, F.; Abraham, J.; et al. Serine Protease HTRA1 as a Novel Target Antigen in Primary Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1666–1681.
- Rosenzwajg, M.; Languille, E.; Debiec, H.; Hygino, J.; Dahan, K.; Simon, T.; Klatzmann, D.; Ronco, P. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int. 2017, 92, 227–237.
- Le, W.-B.; Shi, J.; Zhang, T.; Liu, L.; Qin, H.-Z.; Liang, S.; Zhang, Y.; Zheng, C.-X.; Jiang, S.; Qin, W.-S.; et al. HLA-DRB1*15:01 and HLA-DRB3*02:02 in PLA2R-Related Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 1642–1650.
- Lv, J.; Hou, W.; Zhou, X.-J.; Liu, G.; Zhou, F.; Zhao, N.; Hou, P.; Zhao, M.; Zhang, H. Interaction between PLA2R1 and HLA-DQA1 Variants Associates with Anti-PLA2R Antibodies and Membranous Nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1323–1329.
- Cui, Z.; Xie, L.-J.; Chen, F.-J.; Pei, Z.-Y.; Zhang, L.-J.; Qu, Z.; Huang, J.; Gu, Q.-H.; Zhang, Y.-M.; Wang, X.; et al. MHC Class II Risk Alleles and Amino Acid Residues in Idiopathic Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 1651–1664.
- Xie, J.; Liu, L.; Mladkova, N.; Li, Y.; Ren, H.; Wang, W.; Cui, Z.; Lin, L.; Hu, X.; Yu, X.; et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat. Commun. 2020, 11, 1600.
- Xu, X.; Wang, G.; Chen, N.; Lu, T.; Nie, S.; Xu, G.; Zhang, P.; Luo, Y.; Wang, Y.; Wang, X.; et al. Long-Term Exposure to Air Pollution and Increased Risk of Membranous Nephropathy in China. J. Am. Soc. Nephrol. 2016, 27, 3739–3746.
- Debiec, H.; Valayannopoulos, V.; Boyer, O.; Nöel, L.-H.; Callard, P.; Sarda, H.; De Lonlay, P.; Niaudet, P.; Ronco, P. Allo-Immune Membranous Nephropathy and Recombinant Aryl Sulfatase Replacement Therapy: A Need for Tolerance Induction Therapy. J. Am. Soc. Nephrol. 2013, 25, 675–680.
- Huang, C.C.; Lehman, A.M.; Albawardi, A.; A Satoskar, A.; Brodsky, S.V.; Nadasdy, G.; A Hebert, L.; Rovin, B.H.; Nadasdy, T. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod. Pathol. 2013, 26, 799–805.
- Luo, W.; Olaru, F.; Miner, J.H.; Beck, L.H.J.; Van Der Vlag, J.; Thurman, J.M.; Borza, D.-B. Alternative Pathway Is Essential for Glomerular Complement Activation and Proteinuria in a Mouse Model of Membranous Nephropathy. Front. Immunol. 2018, 9, 1433.
- Tao, M.H.; Canfield, S.M.; Morrison, S.L. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH-terminal sequence of the CH2 domain. J. Exp. Med. 1991, 173, 1025–1028.
- Segawa, Y.; Hisano, S.; Matsushita, M.; Fujita, T.; Hirose, S.; Takeshita, M.; Iwasaki, H. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr. Nephrol. 2010, 25, 1091–1099.
- Brglez, V.; Boyer-Suavet, S.; Seitz-Polski, B. Complement Pathways in Membranous Nephropathy: Complex and Multifactorial. Kidney Int. Rep. 2020, 5, 572–574.
- Haddad, G.; Lorenzen, J.M.; Ma, H.; de Haan, N.; Seeger, H.; Zaghrini, C.; Brandt, S.; Kölling, M.; Wegmann, U.; Kiss, B.; et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1–associated membranous nephropathy. J. Clin. Investig. 2021, 131, e140453.
More
Information
Subjects:
Urology & Nephrology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
31 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No