Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yansheng Du | + 1464 word(s) | 1464 | 2022-02-24 06:55:24 | | | |
| 2 | Ali Zarrabi | -2 word(s) | 1462 | 2022-03-30 03:49:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Du, Y.; Zarrabi, A. Pb Induces MCP-1 in the Choroid Plexus. Encyclopedia. Available online: https://encyclopedia.pub/entry/21130 (accessed on 28 June 2026).
Du Y, Zarrabi A. Pb Induces MCP-1 in the Choroid Plexus. Encyclopedia. Available at: https://encyclopedia.pub/entry/21130. Accessed June 28, 2026.
Du, Yansheng, Ali Zarrabi. "Pb Induces MCP-1 in the Choroid Plexus" Encyclopedia, https://encyclopedia.pub/entry/21130 (accessed June 28, 2026).
Du, Y., & Zarrabi, A. (2022, March 28). Pb Induces MCP-1 in the Choroid Plexus. In Encyclopedia. https://encyclopedia.pub/entry/21130
Du, Yansheng and Ali Zarrabi. "Pb Induces MCP-1 in the Choroid Plexus." Encyclopedia. Web. 28 March, 2022.
Copy Citation
Lead (Pb) is an environmental element that has been implicated in the development of dementia and Alzheimer’s disease (AD). Additionally, innate immune activation contributes to AD pathophysiology. However, the mechanisms involved remain poorly understood. The choroid plexus (CP) is not only the site of cerebrospinal fluid (CSF) production, but also an important location for communication between the circulation and the CSF.
lead
Alzheimer’s disease
MCP-1
choroid plexus
Z310
1. Introduction
The demand for Pb in industry has been steadily increasing during the last decade. In addition to the occupational hazard, environmental exposure to Pb continues to be a major public health concern, as Pb is widely present in the air, drinking water, household products, plastics, and painted materials [1]. While Pb can cause acute toxicity, previous human studies suggested that cumulative lifetime Pb exposure is also associated with accelerated declines in cognition and dementia [2]. Workers exposed to Pb show brain atrophy and behavioral deficits [3][4][5]. A follow-up study examining Pb workers revealed that cumulative Pb-dose-related exposure is associated with progressive declines in cognitive function as well as alterations in brain structure [2]. Additionally, reports show the presence of higher levels of Pb in diffuse neurofibrillary tangles in AD cases compared to control individuals [6][7]. A retrospective human study demonstrated a relationship between prenatal Pb exposure and the alteration of genes and enzymes implicated in AD senile amyloid plaque formation [8]. Cumulative evidence shows Pb exposure induces both amyloid deposition and tau hyperphosphorylation in animal brains [9][10][11][12]. However, despite these findings, it is currently unclear how Pb contributes to the pathogenesis and pathology of AD and related dementia (ADRD). Understanding Pb exposure and its relationship to ADRD is of urgent importance for the broader community. Data from the United States show that those exposed to high levels of Pb in the 1960s and 1970s are now at an age where they are at higher risk of developing AD, with an estimated prevalence of 8.4–13.8 million [13][14].
2. Pb Exposure Induced MCP-1 Expression and Enhanced Macrophage Infiltration in the CP Tissues
After Tg-SwDI mice were treated with or without Pb daily for 30 days, MCP-1 expression levels and macrophage infiltration in mouse CP areas were evaluated by using immunohistochemical assays. As shown in Figure 1A,B, chronic Pb exposure significantly increased expression of MCP-1 in this area. The fluorescence intensity of MCP-1 in the CP increased 1.9-fold compared to the control group (p < 0.001). Additionally, since MCP-1 is a chemoattractant for macrophage migration and infiltration, researchers investigated if induction of MCP-1 was able to affect the macrophage number in the CP. As expected, along with the induction of MCP-1, Pb exposures also increased Iba-1+/CD45+ macrophages in the CP areas. The percentage of Iba1+/CD+ macrophages against total cells markedly increased from 4.8 ± 0.19% to 7.0 ± 0.33% (p < 0.01, Figure 1C,D). Both the NF-κB and p38 MAPK pathways were implicated in the induction of MCP-1 expression [15]. researchers therefore evaluated the fluorescence intensity of phosphorylated NF-κB p65 (p-p65) and phosphorylated p38 MAP kinase (p-p38) in the CP. As expected, either p-p65 or p-p38 MAP kinase increased 1.6-fold (Figure 1E–H) compared to the control group (p < 0.05).
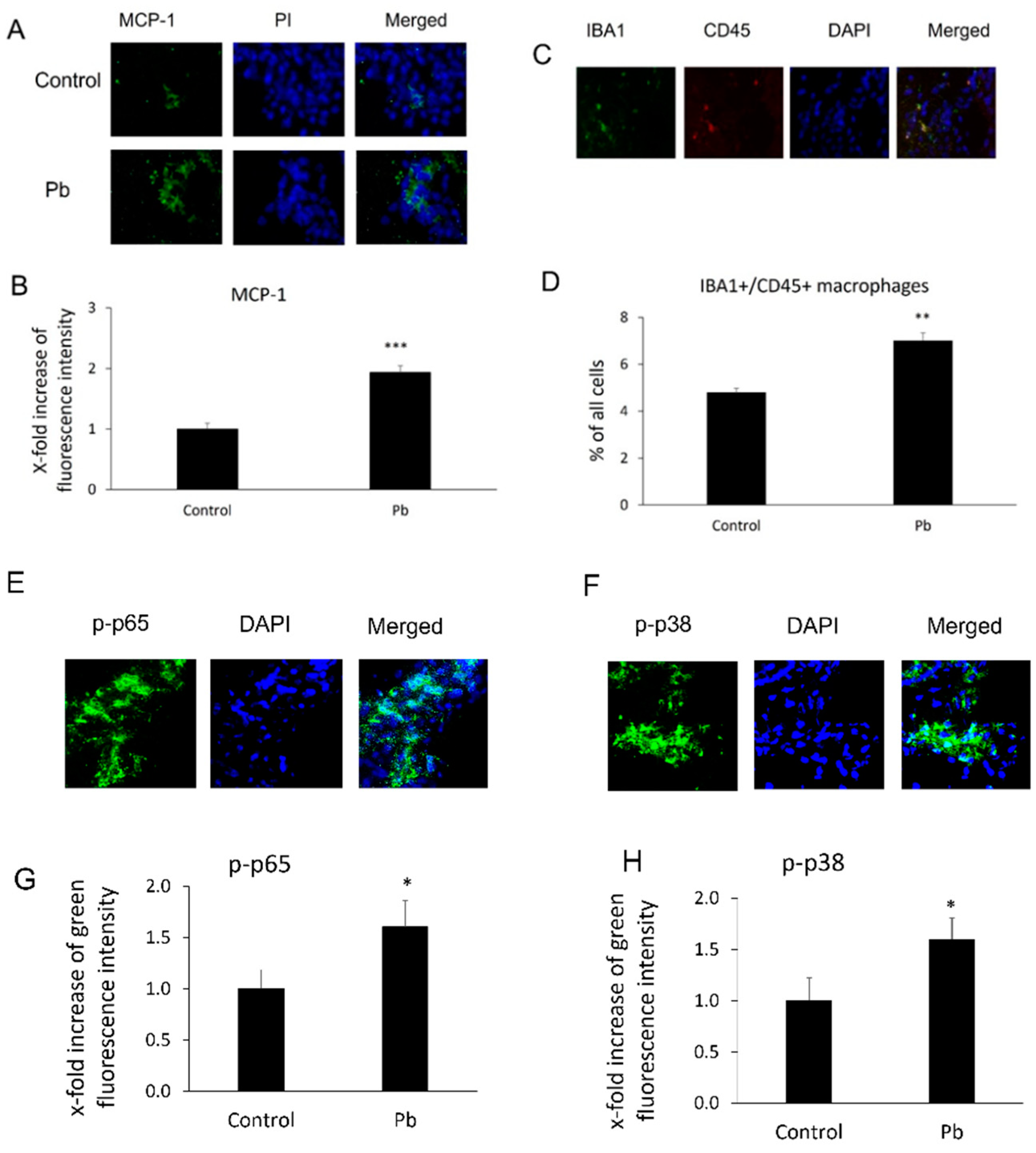
Figure 1. Pb exposure induced MCP-1 expression, phosphorylated NF-κB p65 (p-p65), phosphorylated p38 MAPK (p38), and macrophage infiltration in the CP tissues. Tg-SwDI transgenic mice received oral gavage of 50 mg/kg Pb acetate once daily for 30 days. At the end of Pb exposure, mice brain sections were stained with antibodies of MCP-1, p-p65, and p-p38 to determine MCP-1 expression and levels of p-p65 or p-p38, as well as with both Iba1 and CD45 antibodies to detect macrophage infiltration in the CP. (A) Representative MCP-1 immunofluorescent images used for counting. (B) Quantification of MCP-1 expression by analyzing total MCP-1 fluorescent intensity in the CP. Upregulation of MCP-1 expression in Pb-treated mice was observed compared to that in mice without Pb treatments in the CP. (C) Representative double immunofluorescent images of Iba1+/CD45+ used for counting. (D) Quantification of Iba1+/CD45+ macrophages in the CP. (E) Representative p-p65 immunofluorescent images used for counting. (F) Quantification of p-p65 levels by analyzing total p-p65 fluorescent intensity in the CP. (G) Representative p-p38 immunofluorescent images used for counting. (H) Quantification of p-p38 levels by analyzing total p-p38 fluorescent intensity in the CP. Upregulation of MCP-1 expression, macrophage infiltration, and p-p65 and p-p38 in Pb-treated mice was observed as compared to mice without Pb treatments in the CP. Data are presented as mean ± SD, n = 3/group. * p < 0.05, ** p < 0.01, *** p < 0.001.
3. Pb Exposure Induced MCP-1 Expression in Rat Choroidal Epithelial Z310 Cells
Since the CP epithelial cells are likely the major source of MCP-1 production in the CP, researchers employed immortalized choroidal epithelial Z310 cells to further evaluate in vivo observation that Pb is able to induce MCP-1. As shown in Figure 2, Pb treatments induced MCP-1 expression in a dose-related fashion within 24 h. Pb at 1, 3, and 5 µM markedly induced MCP-1 expression from 185.4 ± 0.58 pg/mg to 208.5 ± 2.14 pg/mg (p < 0.01), 281.9 ± 17.19 pg/mg (p < 0.01), and 361.7± 75.7 pg/mg (p < 0.05) (Figure 2A), respectively. Consistent with the expression data, 1, 3, and 5 µM of Pb also stimulated MCP-1 release into culture media from 30.0 ± 36.06 pg/mL to 55 ± 49.24 pg/mL, 186.7 ± 52.04 pg/mL (p < 0.05), and 636.7 ± 160/73 pg/mL (p < 0.05), respectively (Figure 2B).
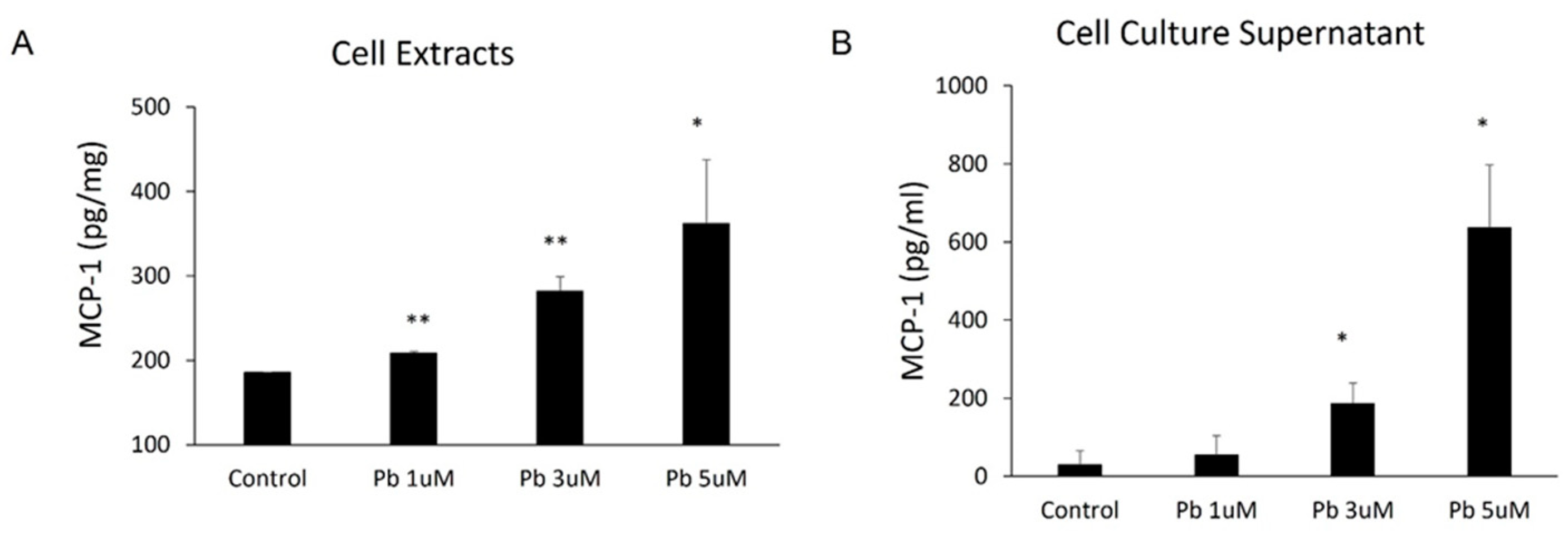
Figure 2. Pb exposure induced MCP-1 expression in rat choroidal epithelial Z310 cells. Z310 cells were treated with 1, 3, or 5 µM Pb for 24 h. MCP-1 levels in both cell extracts (A) and cell-free culture supernatants (B) were determined by using ELISA. Data represent mean ± SD, n = 3/group. *: p < 0.05, **: p < 0.01.
4. Both NF-κB p65 and p38 MAPkinase Were Activated in Z310 Cells by Pb
Based on the in vivo data, researchers decided to investigate whether Pb exposure is able to induce NF-κB p65 or p38 MAPK activation in CP epithelial cells. Phosphorylation levels of both NF-κB p65 and its upstream factor, IKKα, or p38 MAPK and its downstream substrate, ATF-2, were measured in Z310 cells following 1 or 3 h Pb treatments. As shown in Figure 2, there was a 1.8-fold (p < 0.01) increase in phosphor-NF-kB p65 (p-p65) and 2.1-fold (p < 0.05) in phosphor-IKKα (p-IKKα) 1 h after Pb exposure (Figure 3A,C). Additionally, there was a 2.0- (1 h after Pb, p < 0.01) or 2.3-fold (3 h after Pb, p < 0.01) increase in phosphor-p38 MAPK (p-p38, Figure 3B), and a 1.8-fold (3 h after Pb, p < 0.05) increase in phosphor-ATF-2 (p-ATF-2, Figure 3D), suggesting Pb exposure is able to activate both pathways in CP epithelial cells.
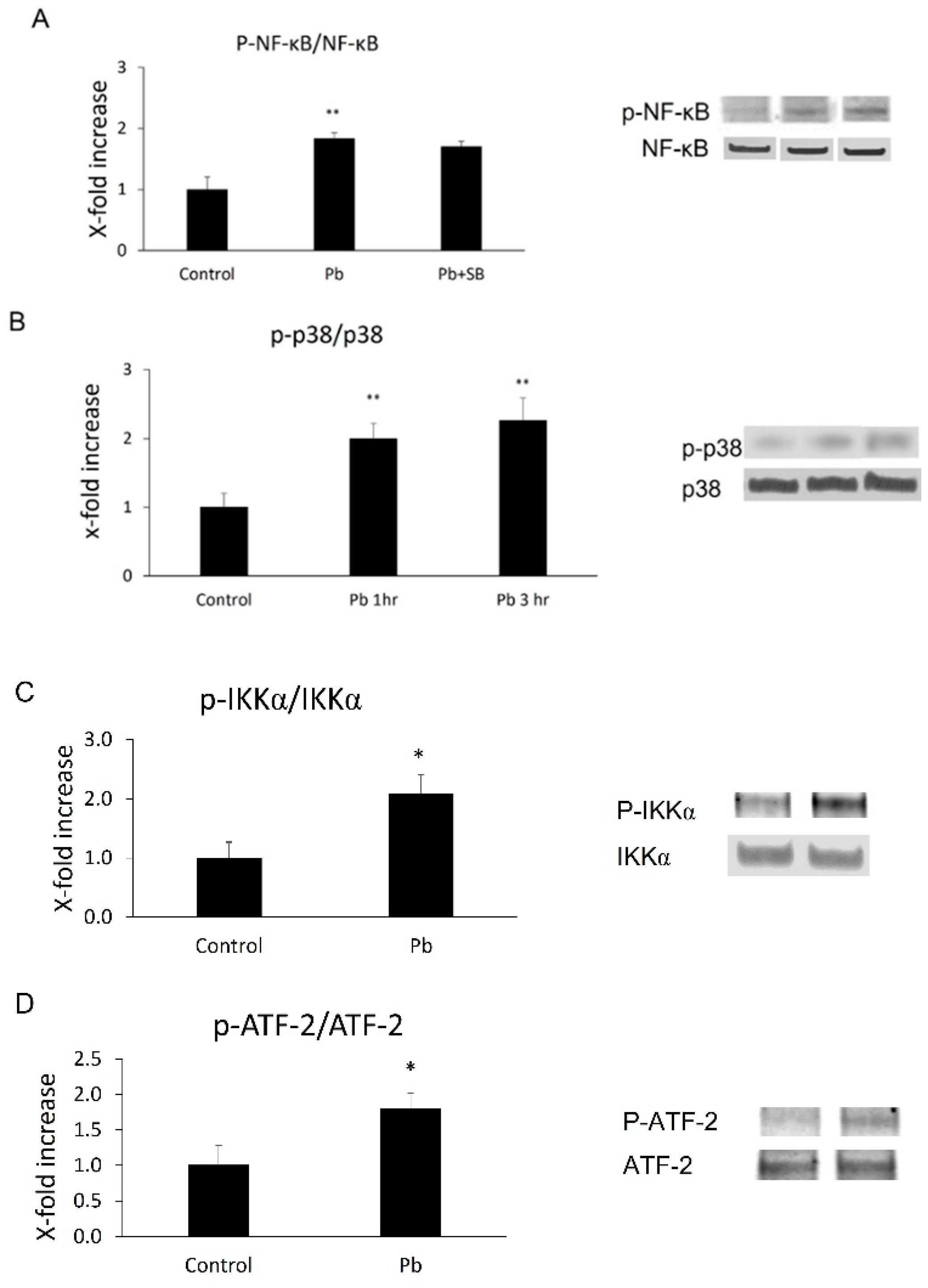
Figure 3. Phosphorylation levels of NF-κB p65, IKKα, p38 MAP kinase, and ATF-2 in Z310 cells followed by Pb treatments were determined by Western blot. (A,C) After 1 h pre-treatments with or without 10 µM SB 203580, Z310 cells were incubated with 1 µM Pb for an additional 1 h. Cells were then lysed and levels of phosphorylated and total NF-κB p65 (p-p65, p65) or IKKα (p-IKKα and IKKα) in Z310 were determined by Western blot. The phosphorylated protein (p-p65 or p-IKKα) band intensities were quantified and normalized to p65 or IKKα densities using Image J. (B,D) Z310 cells were treated with 1 µM Pb for 1 or 3 h. Western blot was performed to determine phosphorylated and total p38 MAPK (p-p38, p38) and ATF-2 (p-ATF-2, ATF-2). The phosphorylation levels of p38 MAPK or ATF-2 (p-p38, p-ATF-2) were also normalized by protein levels of total p38 or ATF-2. Data are presented as mean ± SD, n = 3/group. *: p < 0.05, ** p < 0.01.
5. Both NF-κB and p38 MAPK Inhibitors Blocked Pb-Induced MCP-1 Expression
researchers then investigated whether Pb-induced activations of NF-κB and p38 MAPK underlies Pb-induced MCP-1 expression. As expected, both the NF-κB inhibitor, BAY 11-7082, and the p38 MAPK inhibitor, SB 203580, significantly inhibited Pb-induced MCP-1 expression. MCP-1 expression levels were reduced from 208.5 ± 6.82 pg/mg to 196.6 ± 0.02 pg/mg (p < 0.05) by 1 µM BAY 11-7082 and to 194.0 ± 0.03 pg/mg (p < 0.05) by 10 µM SB 203580 (Figure 4). Thus, the data show that Pb induces the expression of MCP-1 in the CP epithelial cells via both Pb-induced NF-κB p65 and p38 MAPK pathways.
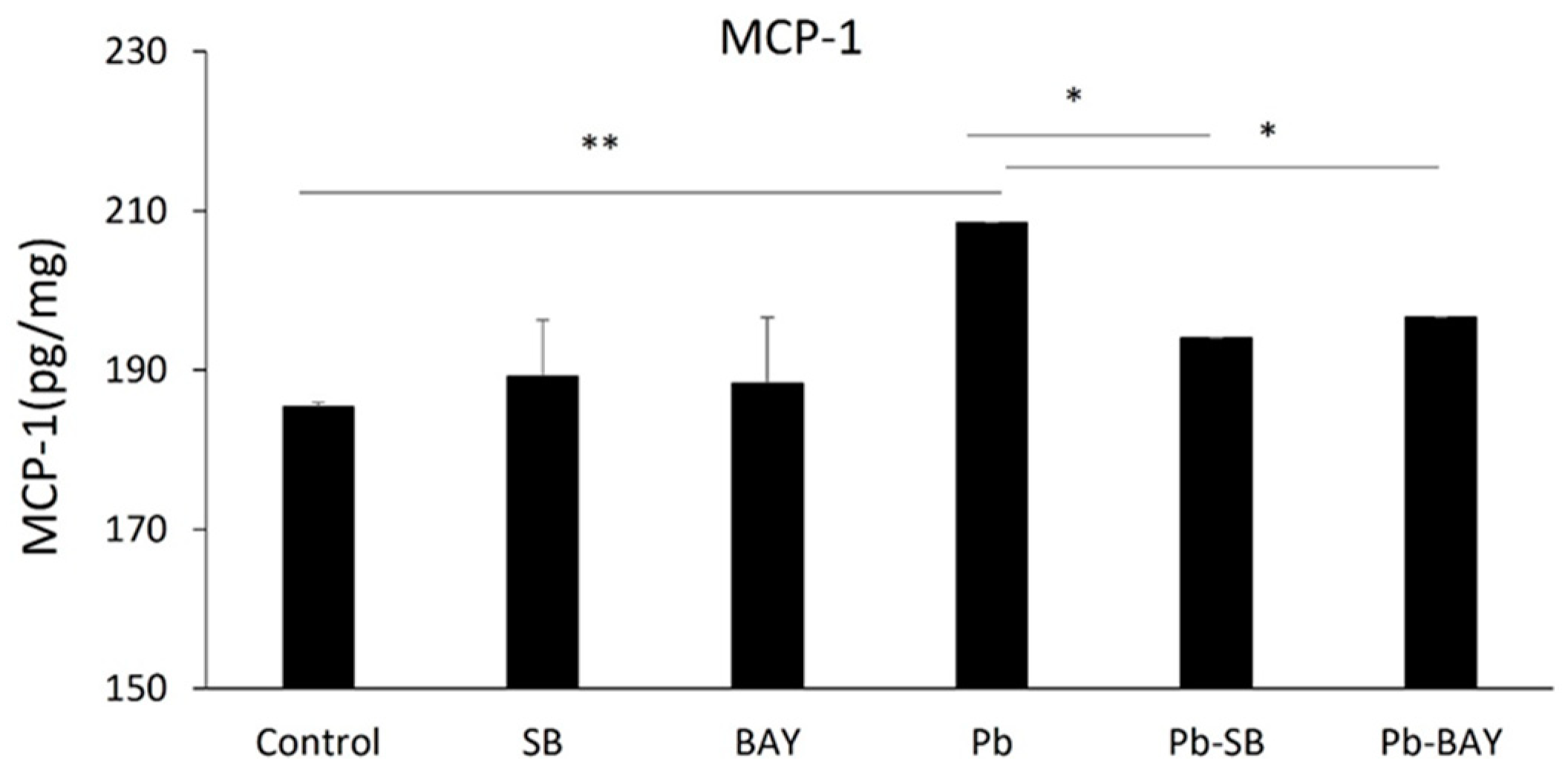
Figure 4. Both NF-κB and p38 MAPK inhibitors blocked Pb-induced MCP-1 expression. Z310 cells were pre-treated with/without 10 µM SB 203580 or 1 µM BAY 11-7082 for 1 h, followed by 1 µM Pb treatments for additional 24 h. Cells were lysed and expression levels of MCP-1 in cell extracts were quantified using ELISA. Data are presented as mean ± SD, n = 3/group. * p < 0.05, ** p < 0.01.
6. SB 253,580 Did Not Block NF-κB p65 Phosphorylation
It remains unclear if these two pathways individually or together mediate Pb-induced MCP-1 expression. It was reported that the p38 MAPK pathway does not directly mediate NiCl-induced NF-κB p65 phosphorylation in primary endothelial cells [16] during MCP-1 induction, but targets the downstream factor of NF-κB. Similar to that case, researchers observed that SB 203580 did not block Pb-induced NF-κB p65 phosphorylation (Figure 3A), suggesting Pb might at least inhibit the downstream target of NF-κB.
References
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.F. Brain barrier systems: A new frontier in metal neurotoxicological research. Toxicol. Appl. Pharm. 2003, 192, 1–11.
- Bakulski, K.M.; Rozek, L.S.; Dolinoy, D.C.; Paulson, H.L.; Hu, H. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr. Alzheimer Res. 2012, 9, 563–573.
- Stewart, W.F.; Schwartz, B.S.; Simon, D.; Kelsey, K.; Todd, A.C. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ. Health Perspect. 2002, 110, 501–505.
- Stewart, W.F.; Schwartz, B.S.; Davatzikos, C.; Shen, D.; Liu, D.; Wu, X.; Todd, A.C.; Shi, W.; Bassett, S.; Youssem, D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology 2006, 66, 1476–1484.
- Schwartz, B.S.; Caffo, B.; Stewart, W.F.; Hedlin, H.; James, B.D.; Yousem, D.; Davatzikos, C. Evaluation of cumulative lead dose and longitudinal changes in structural magnetic resonance imaging in former organolead workers. J. Occup. Environ. Med. 2010, 52, 407–414.
- Graves, A.B.; van Duijn, C.M.; Chandra, V.; Fratiglioni, L.; Heyman, A.; Jorm, A.F.; Kokmen, E.; Kondo, K.; Mortimer, J.A.; Rocca, W.A.; et al. Occupational exposures to solvents and lead as risk factors for Alzheimer’s disease: A collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 1991, 20 (Suppl. 2), S58–S61.
- Haraguchi, T.; Ishizu, H.; Takehisa, Y.; Kawai, K.; Yokota, O.; Terada, S.; Tsuchiya, K.; Ikeda, K.; Morita, K.; Horike, T.; et al. Lead content of brain tissue in diffuse neurofibrillary tangles with calcification (DNTC): The possibility of lead neurotoxicity. Neuroreport 2001, 12, 3887–3890.
- Mazumdar, M.; Xia, W.; Hofmann, O.; Gregas, M.; Ho Sui, S.; Hide, W.; Yang, T.; Needleman, H.L.; Bellinger, D.C. Prenatal lead levels, plasma amyloid beta levels, and gene expression in young adulthood. Environ. Health Perspect. 2012, 120, 702–707.
- Basha, M.R.; Murali, M.; Siddiqi, H.K.; Ghosal, K.; Siddiqi, O.K.; Lashuel, H.A.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. Lead (Pb) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005, 19, 2083–2084.
- Gu, H.; Robison, G.; Hong, L.; Barrea, R.; Wei, X.; Farlow, M.R.; Pushkar, Y.N.; Du, Y.; Zheng, W. Increased beta-amyloid deposition in Tg-SWDI transgenic mouse brain following in vivo lead exposure. Toxicol. Lett. 2012, 213, 211–219.
- Zhang, J.; Cai, T.; Zhao, F.; Yao, T.; Chen, Y.; Liu, X.; Luo, W.; Chen, J. The role of alpha-synuclein and tau hyperphosphorylation-mediated autophagy and apoptosis in lead-induced learning and memory injury. Int. J. Biol. Sci. 2012, 8, 935–944.
- Bihaqi, S.W.; Bahmani, A.; Adem, A.; Zawia, N.H. Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: Relevance to AD. Neurotoxicology 2014, 44, 114–120.
- Juracek, K.E.; Ziegler, A.C. The legacy of leaded gasoline in bottom sediment of small rural reservoirs. J. Environ. Qual. 2006, 35, 2092–2102.
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783.
- Zhong, Y.; Liu, T.; Guo, Z. Curcumin inhibits ox-LDL-induced MCP-1 expression by suppressing the p38MAPK and NF-kappaB pathways in rat vascular smooth muscle cells. Inflamm. Res. 2012, 61, 61–67.
- Goebeler, M.; Gillitzer, R.; Kilian, K.; Utzel, K.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood 2001, 97, 46–55.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
632
Revisions:
2 times
(View History)
Update Date:
30 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No