Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cynthia L. Monaco | + 2634 word(s) | 2634 | 2022-02-25 06:38:10 | | | |
| 2 | Dean Liu | Meta information modification | 2634 | 2022-03-30 03:19:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Monaco, C. Transkingdom Analysis of the Female Reproductive Tract. Encyclopedia. Available online: https://encyclopedia.pub/entry/21121 (accessed on 25 June 2026).
Monaco C. Transkingdom Analysis of the Female Reproductive Tract. Encyclopedia. Available at: https://encyclopedia.pub/entry/21121. Accessed June 25, 2026.
Monaco, Cynthia. "Transkingdom Analysis of the Female Reproductive Tract" Encyclopedia, https://encyclopedia.pub/entry/21121 (accessed June 25, 2026).
Monaco, C. (2022, March 28). Transkingdom Analysis of the Female Reproductive Tract. In Encyclopedia. https://encyclopedia.pub/entry/21121
Monaco, Cynthia. "Transkingdom Analysis of the Female Reproductive Tract." Encyclopedia. Web. 28 March, 2022.
Copy Citation
The female reproductive tract (FRT) microbiome plays a vital role in maintaining vaginal health. Viruses are key regulators of other microbial ecosystems, but little is known about how the FRT viruses (virome), particularly bacteriophages that comprise the phageome, impact FRT health and dysbiosis.
virome
microbiome
bacterial vaginosis
bacteriophage
1. Introduction
The female reproductive tract (FRT) houses a compositionally dynamic environment where the host participates in an intricate interplay with a microbiome composed of bacteria and archaea (bacteriome), fungi (fungome), viruses (virome), and occasional protozoal parasites [1][2]. The FRT microbiome plays an important protective role in maintaining vaginal health and preventing urogenital diseases such as bacterial vaginosis (BV), yeast infections, pre-term birth, and sexually transmitted infections (STIs), including human immunodeficiency virus (HIV) [3][4][5][6]. Prior studies of the FRT microbiome have primarily focused on determining bacterial composition and function. At least five different bacterial community groupings have been described within the FRT, distinguishable by the dominance of Lactobacillus species or the presence of more diverse anaerobes [7][8]. The prevalence of these communities varies by race and ethnic group, with the majority of Caucasian women hosting Lactobacillus-dominant FRT microbiomes, whereas African women tend to be asymptomatically colonized by higher diversity FRT microbiota [8][9]. Lactobacillus-dominant FRT bacteriomes, especially L. crispatus, protect against vaginal disease through several mechanisms, including by competitive exclusion of pathogenic bacteria for space and nutrients, promoting an acidic vaginal environment via production of lactic acid, and maintaining a low inflammatory state [10][11][12]. BV, the most common cause of vaginal discharge in reproductive age women, is a symptomatic clinical condition characterized by a shift in the FRT microbiota away from a low inflammatory, Lactobacillus-dominant bacteriome to a more diverse community including facultative anaerobes. BV is associated with an increased risk of STI acquisition and pre-term birth [13][14]. Specific BV-associated bacteria include Gardnerella vaginalis, Prevotella, Fusobacterium, Atopobium vaginae, Megasphaera, and Sneathia, among others [15]. FRT bacteriome shifts can occur rapidly and may be related to shifts in bacteriophage populations [16][17].
2. The FRT Bacteriome Clusters into Community Groups
Prior studies examining Western cohorts have identified five bacterial community state types (CSTs), of which type I–III and V are Lactobacillus-dominant while CST IV is comprised of polymicrobial communities [8]. However, studies examining the FRT bacteriome in African cohorts have revealed a different pattern in hierarchical clustering analysis, with more community groupings of high diversity bacteriomes, consistent with the increased prevalence of high diversity FRT bacteriomes in this population [4][8][18]. To further assess the bacterial communities within the FRT bacteriome seen in African women, 253 vaginal swabs were processed and underwent 16S rRNA gene amplicon sequencing of the V3–V4 region [19]. A total of 11 samples did not amplify, and 4 failed to achieve sufficient reads for downstream analysis, leaving 238 samples for bacteriome analysis and sequence identification using QIIME2 (Figure 1A).

Figure 1. Bacteriome profiling by community group of self-collected vaginal swabs from South African women. (A) Relative abundance of the 16 most frequently identified bacterial taxa (y-axis) by sample (x-axis), grouped by community group (CG), bacterial vaginosis (BV) status, visit number, highly active antiretroviral therapy (HAART) status, and human immunodeficiency virus (HIV) status (color key shown). Percent abundance is indicated by gradient key. Using Ward’s linkage hierarchical clustering, samples clustered into five distinct bacterial community profiles termed CG. (B) Bacterial composition (color key shown) for each of the five CGs (x-axis) expressed as relative abundance (y-axis). (C) Bar plot showing the relative abundance of 16S rRNA copies per 10 ng total DNA (y-axis) of L. iners (blue), L. crispatus (purple), L. gasseri (pink), and L. jensenii (green) bacterial species as determined by qPCR of FRT samples (x-axis) that clustered into CG1.
The 16S samples were analyzed using VALENCIA [20], a program developed to assign 16S vaginal samples to the commonly used CST communities [8] and employs similarity scores ranging from 0 (no shared taxa) to 1 (all taxa shared and at the same relative abundance) to assess assignment confidence. VALENCIA successfully assigned L. iners-dominant samples to CST III (L. iners-dominant CST) with high confidence (similarity score 91%). However, the remaining CST assignments were low confidence with an overall similarity score average of 29.7%. This likely reflects the bias of VALENCIA toward Lactobacillus-dominant samples. Because samples within this cohort were predominantly high diversity, Ward’s linkage hierarchal clustering was used to classify samples into distinct bacterial communities by composition and relative abundance.
Hierarchical clustering analysis of all visits by community abundance and composition identified five unique bacterial community clusters named herein bacterial community groups (CGs), which were distinguished by Lactobacillus-dominance (CG1 and 2) or higher diversity bacteriomes (CG3–5; Figure 1A). Similar to other African cohorts [4][8][18], the majority (n = 173, 72.7%) of subjects had high diversity FRT bacteriomes with a low prevalence of Lactobacillus-dominant bacteriomes. Samples that were dominated by a single species, defined as >50% community composition, made up 42.0% of all samples and were mostly clustered with CG1 and 2, while the remaining samples showed no individual dominant species and mainly clustered in CG3–5. CG1 (n = 15, 6.3%; Figure 1B), a low diversity FRT bacteriome, was comprised almost exclusively of Lactobacillus species (78%) that were unable to be further delineated by 16S rRNA gene amplicon sequencing. To further define the predominant Lactobacillus constituents of CG1, qPCR of 16S rRNA gene sequences from the key FRT Lactobacillus species L. iners, L. crispatus, L. gasseri, and L. jensenii [9][18] was performed, revealing that approximately 50% of samples in CG1 were L. crispatus-dominant, similar to what has been previously described as CST I [8] (Figure 1C). One vaginal swab in CG1 contained sufficient volume to only perform qPCR for L. iners and L. crispatus, and L. crispatus was most abundant (not shown). L. jensenii tended to predominate when present (Figure 1C). CG2 (n = 54, 22.7%) was L. iners-dominant, with a few samples showing notable amounts of Gardnerella and Prevotella as well (Figure 1B). The identified high diversity CGs 3–5 were compositionally different from the conventional CST4 subtypes [8]. The second to largest and most diverse group, CG3 (n = 64, 26.9%), consisted mainly of Gardnerella, Prevotella, and L. iners (Figure 1B). The smallest high diversity CG was CG4 (n = 30, 12.6%), in which Shuttleworthia and Gardnerella were predominant. CG5 contained the largest number of samples (n = 75, 31.5%) and was dominated by Sneathia and Prevotella (Figure 1B). CG3, CG4 and CG5 exhibited significantly higher alpha diversity than CG1 and 2 (Figure 2A; p = 0.0001, 0.0065, and <0.0001, respectively). Beta diversity significantly differed between these five bacterial CGs (Figure 2B; p = 0.0001).

Figure 2. FRT bacteriome clusters into distinct community groups (CGs) that differ by alpha and beta diversity. (A) Bacterial Shannon diversity (y-axis) by CG (x-axis) as determined by linear mixed-effects model. Center bar represents median; gray box bounded by upper/lower interquartile ranges (IQR); whiskers represent range; dots represent outliers; color-filled areas are representative of density/distribution of diversity values. **, p < 0.01; ***, p < 0.001. (B) Principal coordinate analysis (PCoA) plot of the weighted UniFrac distances colored by CG (color key shown).
3. Bacteriophages Comprise the Majority of the FRT DNA Virome
While the FRT bacteriome has been well-studied, the FRT virome, especially bacteriophage populations, is relatively unknown. Therefore, the FRT DNA virome was characterized using a subset of baseline samples by enriching for VLPs from resuspended vaginal swabs and extracting viral nucleic acid [21]. Libraries were constructed and sequenced using the Illumina NovaSeq platform for a subset of 38 baseline samples, 14 of which were BV-negative and 24 were BV-positive. The resulting viral sequences underwent quality control, removal of bacterial and human reads, and then were assigned to known viral taxa using VirusSeeker, a BLAST-based NGS virome analysis pipeline [22]. On average, there were 29 million reads per sample, 86.8% of them being high quality, with approximately 58% unique reads per sample.
The DNA eukaryotic virome was comprised almost entirely of Papillomaviridae (94% of eukaryotic viral reads). Herpesviridae (including herpes simplex virus, cytomegalovirus, and Epstein Barr virus) comprised 5% of viral reads, while Polyomaviridae, Poxiviridae (mulluscum contagiosum virus), and Anelloviridae combined were less than 1%. There was no significant difference in reads assigned to DNA eukaryotic viruses by BV or HIV status after correction for multiple comparisons. However, FRT bacteriophages were abundant, representing 83% of virally assigned reads. Samples contained sequences identified as belonging to the Myoviridae, Siphoviridae, Podoviridae, Inoviridae, Ackermannviridae, Microviridae, Lipothrixviridae, Plasmaviridae, and Tectiviridae bacteriophage families. Sequences assigned to members of the Caudovirales order, lytic-tailed dsDNA bacteriophages, including Myoviridae, Siphoviridae, and Podoviridae, were the most abundant in all samples regardless of BV, HAART, or HIV status.
Bacteriophages within the FRT are clustered into two distinct, novel bacteriophage community groups based on composition and abundance that were named viral state types (VSTs; Figure 3A). VST1 represented 44.7% (n = 17) of all samples while VST2 contained the remaining 55.3% (n = 21). Bacteriophage Shannon diversity differed between VSTs (Figure 3B), with VST2 having the highest diversity bacteriophage populations. The VSTs were also grouped distinctly by beta diversity analysis (Figure 3C; PERMANOVA p = 0.0001). Neither VST exhibited a dominant bacteriophage member. VST1 contained several samples with a high relative abundance of Rhodococcus viruses, Spounavirinae, phi29 virus and Biseptimavirus-assigned bacteriophage reads. VST2 composition was more evenly distributed. This is the first study to identify bacteriophage community groups in the FRT.
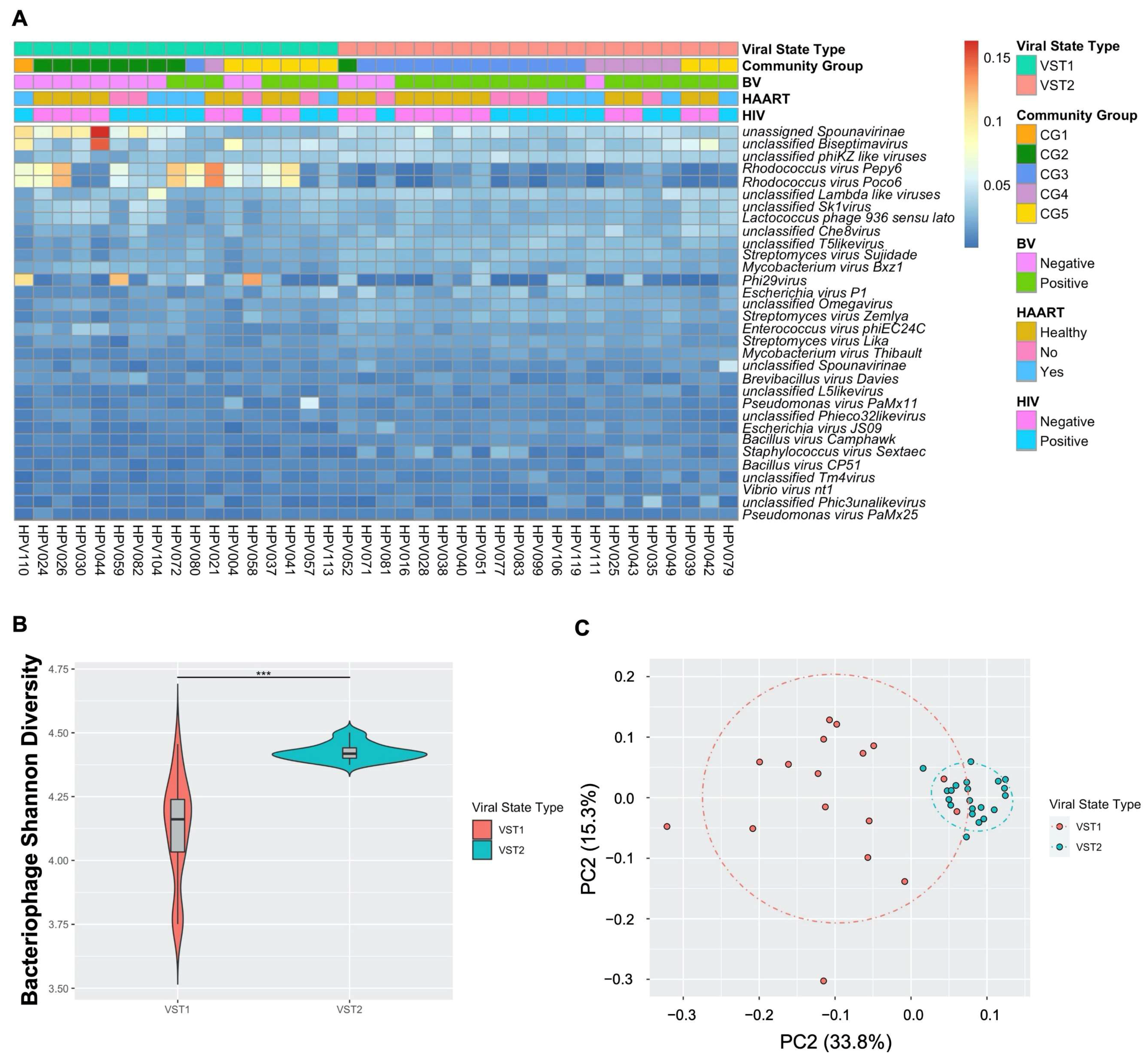
Figure 3. FRT DNA bacteriophages cluster into two unique community groups. Self-collected vaginal swabs were processed for DNA virome analysis by enriching for viral nucleic acid, libraries built and sequenced. (A) Relative abundance of the 32 most frequent bacteriophage species (y-axis) by sample (x-axis). Ward’s linkage hierarchical clustering analysis was used to cluster samples into distinct bacteriophage community profiles called viral state types (VSTs). VST, bacterial vaginosis (BV) status, highly active antiretroviral therapy (HAART) status, and human immunodeficiency virus (HIV) status (color key) are shown. Percent abundance is indicated by gradient key. (B) Bacteriophage Shannon diversity (y-axis) by VST (x-axis) as determined by linear regression model. Center bar represents median; gray box is bounded by upper/lower interquartile ranges (IQR); whiskers represent range; dots represent outliers; color-filled areas are representative of density/distribution of diversity values. ***, p < 0.001. (C) Principal coordinate analysis (PCoA) plots of beta diversity distances, as determined by permutational multivariate analysis of variance, colored by VST.
4. Transkingdom Associations within the FRT Microbiome of South African Women
Bacteriophages can directly impact bacterial composition and abundance through infection of their host. Therefore, the transkingdom associations between bacteriophage and bacteria in the FRT were investigated. The VSTs significantly correlated with bacterial CG (p = 0.00015; Figure 3A), with VST1 associating with the Lactobacillus-dominant CG1 and 2, while VST2 associated with CG3 and 4, both higher diversity CG. CG5 contained samples belonging to both VSTs (Figure 3A). These data indicate a strong association between bacteriophage communities and the host bacterial populations.
The transkingdom interplay between bacteriophage and bacteria in the FRT was further supported by a significant correlation identified between bacteriophage and bacterial alpha diversity (p = 0.00032). To further investigate specific bacteriophage-bacterial interactions, correlations between FRT bacterial and bacteriophage composition were identified by Kendell’s rank correlation coefficient. Reads assigned to bacteriophages Bacillus virus Camphawk and Bacillus virus Pony, which infect members of the Bacillus genus, positively correlated with the BV-associated bacteria Gardnerella, A. vaginae, Prevotella, Sneathia, and Dialister (Figure 4). Reads assigned to unclassified bacteriophages of the E125 genus are also positively associated with Gardnerella and A. vaginae (Figure 4). A number of Bacillus-infecting bacteriophages, including Bacillus virus Pony and Bacillus virus Staley, were inversely associated with bacteria protective from BV, particularly L. iners (Figure 4). Interestingly, in this cohort, bacteriophage associations revealed that Veillonella more closely grouped with Lactobacillus rather than BV-associated bacteria despite the known role of Veillonella in lactose fermentation and higher diversity FRT microbiomes [8]. These data suggest that bacteriophages directly or indirectly interact with FRT bacterial populations in disease states and identify putative FRT bacteriophage-host networks that may play a role in the development and maintenance of BV.
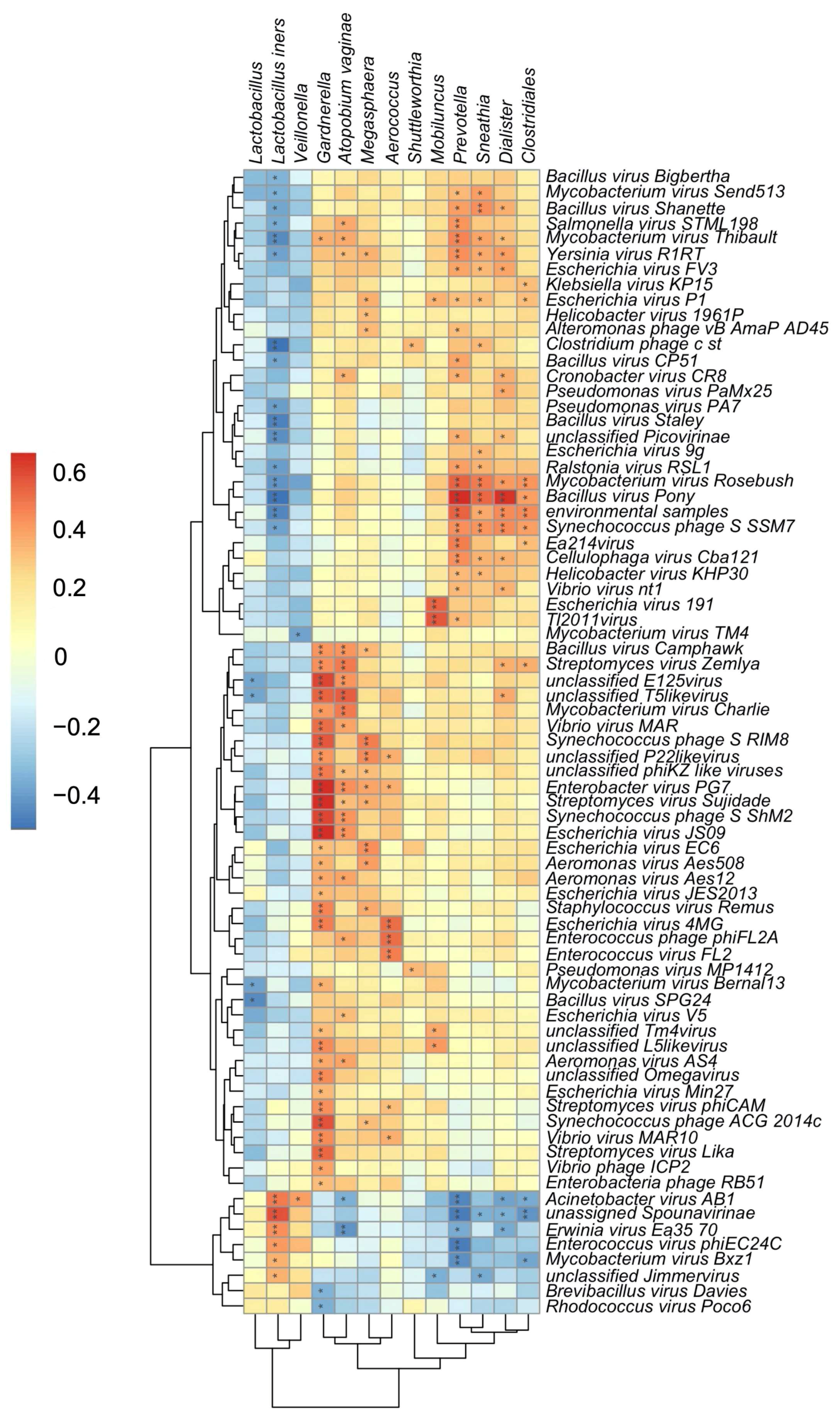
Figure 4. Transkingdom associations within the FRT of South African women. Heatmap of estimated Kendall’s correlation coefficients between FRT bacterial taxa (x-axis) and sequences assigned to bacteriophage (y-axis). Asterix indicate significant correlations after multiple comparisons correction by the Benjamini–Hochberg procedure, *, p < 0.05; **, p < 0.01. Magnitude and sign of the Kendall’s rank correlation coefficient are indicated by gradient key. Red indicates positive correlations; blue indicates negative correlations.
5. Effects of Bacterial Vaginosis on the FRT Virome
Next, the impact of different disease states on this cohort was examined. BV is a clinically significant condition with high morbidity characterized by high bacterial diversity [13][14]. Data showed significant associations between high diversity bacterial CGs and VSTs, and further suggested specific bacteriophage interactions with BV-associated bacteria. Similar to published cohorts [2], BV in this cohort was positively associated with increased bacterial alpha diversity compared to healthy subjects (p = 0.0001). The high diversity CG3–5 (p = 0.0001) also correlated positively with BV. Bacterial taxa, including Gardnerella, Prevotella, Sneathia, and Megasphaera (Figure 5A), varied significantly by clinical BV status, corroborating distinct bacterial signatures in BV. Identification of specific bacterial taxa associated with recovery and transition to BV that could be predictive of the dynamic changes in bacteriome profiles between health and BV were sought. A significant increase in relative abundance of the Gram-positive, facultatively anaerobic cocci Aerococcus (p = 0.00216) and Gemella (p = 0.00746) among participants who recovered from or transitioned to BV was observed, suggesting these may be useful clinical biomarkers of impending change in BV status. While these bacteria are less frequently detected in BV [23], they may represent regulators of FRT bacteriome structure in health and BV.

Figure 5. Discriminant FRT bacterial and bacteriophage species associated with bacterial vaginosis. (A) Significantly discriminant bacterial taxa by BV status were determined by univariate analysis using mixed-effects models. Relative abundance of each taxon (y-axis) is shown in BV-negative (orange) and BV-positive subjects (green; x-axis). All bacterial taxa presented are significant with an FDR-adjusted p < 0.05. (B) Bacteriophage Shannon diversity (y-axis) by BV status (x-axis) as determined by a linear regression model. *, p < 0.05. (C) Grouped bar plot showing the number of identified lytic and lysogenic bacteriophage contigs per sample by BV status. Lytic genes are represented as black triangles and lysogenic genes as black circles. Significance assessed using Mann–Whitney test with multiple comparisons correction by the Benjamini–Hochberg procedure. ***, p < 0.001; ****, p < 0.0001. (D) Significantly discriminant bacteriophage species by clinical BV diagnosis was determined by univariate analysis using a linear regression model. All bacteriophage taxa presented are significant with an FDR-adjusted p < 0.05.
Since clinical BV was associated with significant changes in FRT bacterial composition and correlated with bacterial CGs, and bacteriophage VSTs also correlated with bacterial CGs, correlations between bacteriophage populations and clinical BV were determined. Regression analysis accounting for HIV status and VST revealed bacteriophage Shannon diversity significantly differed by clinical BV diagnosis (Figure 5B; p = 0.0193). To further investigate the manner in which bacteriophage could be affecting bacterial populations in BV, assembled contigs were assessed for the presence of lytic or lysogenic bacteriophage genes to determine bacteriophage lifestyle (Figure 5C). BV-positive samples had significantly more contigs per sample containing lytic (p = 0.000283) and lysogenic (p = 0.000283) genes compared to BV-negative samples (Figure 5C). Moreover, in both BV-positive and BV-negative participants, there were considerably more contigs containing genes associated with a lysogenic lifecycle than those identified as lytic (Figure 5C). Specific bacteriophage taxa differentially abundant in the FRT of BV-positive and -negative women were then ascertained. Bacillus-infecting bacteriophages are known to belong to the Herelleviridae and Podoviridae families [24][25]. Members of these families, including Bacillus virus Camphawk and Bacillus virus Pony (Figure 5D), were significantly associated with BV diagnosis (p= 0.019058 and p = 0.014546, respectively). BV diagnosis also strongly correlated with the bacteriophages Escherichia virus FV3 and unclassified E125 virus (Figure 5D), which are known to infect the BV-associated bacteria Escherichia coli and Burkholderia, respectively [26][27]. Together, these data uncover a link between highly diverse FRT bacteriophage populations, a distinct subset of bacterial hosts, and BV.
6. The Effect of HIV and HPV on the FRT Microbiome
The effect of HIV on the FRT microbiome was also examined in this cohort. No significant difference in bacterial richness, alpha or beta diversity between HIV-positive and HIV-negative subjects was found. Further, there were no significant alterations in bacteriome or bacteriophage diversity by HIV status. Thus, FRT bacterial and bacteriophage communities were not detectably altered by HIV infection in this cohort, suggesting that localized infections may play a more important role in the FRT composition than systemic infections. The relationship between HPV, the main eukaryotic virus found, and bacterial populations were also assessed. Upon examination of 63 HPV-positive and 37 HPV-negative baseline samples, there were no significant associations between HPV infection and bacteriophage or bacterial diversity. Analysis of HPV subtypes revealed increased bacterial alpha diversity in HPV6-positive subjects (p = 0.0181), suggesting certain HPV subtypes may directly or indirectly benefit from the presence of higher diversity bacterial populations, although this analysis may have been underpowered for less prevalent subtypes.
References
- Madere, F.S.; Monaco, C.L. The female reproductive tract virome: Understanding the dynamic role of viruses in gynecological health and disease. Curr. Opin. Virol. 2021, 52, 15–23.
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095.
- Petrova, M.I.; van den Broek, M.; Balzarini, J.; Vanderleyden, J.; Lebeer, S. Vaginal microbiota and its role in HIV transmission and infection. FEMS Microbiol. Rev. 2013, 37, 762–792.
- Gosmann, C.; Anahtar, M.N.; Handley, S.A.; Farcasanu, M.; Abu-Ali, G.; Bowman, B.A.; Padavattan, N.; Desai, C.; Droit, L.; Moodley, A.; et al. Lactobacillus-Deficient Cervicovaginal Bacterial Communities Are Associated with Increased HIV Acquisition in Young South African Women. Immunity 2017, 46, 29–37.
- Greenbaum, S.; Greenbaum, G.; Moran-Gilad, J.; Weintraub, A.Y. Ecological dynamics of the vaginal microbiome in relation to health and disease. Am. J. Obstet. Gynecol. 2019, 220, 324–335.
- Punzón-Jiménez, P.; Labarta, E. The impact of the female genital tract microbiome in women health and reproduction: A review. J. Assist. Reprod. Genet. 2021, 38, 2519–2541.
- Brooks, J.P.; Buck, G.A.; Chen, G.; Diao, L.; Edwards, D.J.; Fettweis, J.M.; Huzurbazar, S.; Rakitin, A.; Satten, G.A.; Smirnova, E.; et al. Changes in vaginal community state types reflect major shifts in the microbiome. Microb. Ecol. Health Dis. 2017, 28, 1303265.
- Smith, S.B.; Ravel, J. The vaginal microbiota, host defence and reproductive physiology. J. Physiol. 2017, 595, 451–463.
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4680–4687.
- Nardis, C.; Mosca, L.; Mastromarino, P. Vaginal microbiota and viral sexually transmitted diseases. Ann. Ig. 2013, 25, 443–456.
- Schellenberg, J.J.; Plummer, F.A. The Microbiological Context of HIV Resistance: Vaginal Microbiota and Mucosal Inflammation at the Viral Point of Entry. Int. J. Inflamm. 2012, 2012, 131243.
- White, D.W.; Keppel, C.R.; Schneider, S.E.; Reese, T.; Coder, J.; Payton, J.E.; Ley, T.J.; Virgin, H.; Fehniger, T.A. Latent herpesvirus infection arms NK cells. Blood 2010, 115, 4377–4383.
- Joesoef, M.R.; Schmid, G. Bacterial vaginosis. Clin. Evid. 2005, 13, 1968–1978.
- Bilardi, J.E.; Walker, S.; Temple-Smith, M.; McNair, R.; Mooney-Somers, J.; Bellhouse, C.; Fairley, C.K.; Chen, M.Y.; Bradshaw, C. The burden of bacterial vaginosis: Women’s experience of the physical, emotional, sexual and social impact of living with recurrent bacterial vaginosis. PLoS ONE 2013, 8, e74378.
- Ngugi, B.M.; Hemmerling, A.; Bukusi, E.A.; Kikuvi, G.; Gikunju, J.; Shiboski, S.; Fredricks, D.N.; Cohen, C.R. Effects of bacterial vaginosis-associated bacteria and sexual intercourse on vaginal colonization with the probiotic Lactobacillus crispatus CTV-05. Sex. Transm. Dis. 2011, 38, 1020–1027.
- Srinivasan, S.; Liu, C.; Mitchell, C.M.; Fiedler, T.L.; Thomas, K.K.; Agnew, K.J.; Marrazzo, J.; Fredricks, D.N. Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PLoS ONE 2010, 5, e10197.
- Brotman, R.M.; Ravel, J.; Cone, R.A.; Zenilman, J.M. Rapid fluctuation of the vaginal microbiota measured by Gram stain analysis. Sex. Transm. Infect. 2010, 86, 297–302.
- Anahtar, M.N.; Byrne, E.H.; Doherty, K.E.; Bowman, B.A.; Yamamoto, H.S.; Soumillon, M.; Padavattan, N.; Ismail, N.; Moodley, A.; Sabatini, M.E.; et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 2015, 42, 965–976.
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6.
- France, M.T.; Ma, B.; Gajer, P.; Brown, S.; Humphrys, M.S.; Holm, J.B.; Waetjen, L.E.; Brotman, R.M.; Ravel, J. VALENCIA: A nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 2020, 8, 166.
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322.
- Zhao, G.; Wu, G.; Lim, E.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30.
- Coleman, J.S.; Gaydos, C.A. Molecular Diagnosis of Bacterial Vaginosis: An Update. J. Clin. Microbiol. 2018, 56, e00342-18.
- Ritz, M.P.; Perl, A.L.; Colquhoun, J.M.; Chamakura, K.R.; Kuty Everett, G.F. Complete Genome of Bacillus subtilis Myophage CampHawk. Genome Announc. 2013, 1, e00984-13.
- Grose, J.H.; Jensen, G.L.; Burnett, S.H.; Breakwell, D.P. Correction: Genomic comparison of 93 Bacillus phages reveals 12 clusters, 14 singletons and remarkable diversity. BMC Genom. 2014, 15, 1184.
- Xia, Q.; Cheng, L.; Zhang, H.; Sun, S.; Liu, F.; Li, H.; Yuan, J.; Liu, Z.; Diao, Y. Identification of vaginal bacteria diversity and its association with clinically diagnosed bacterial vaginosis by denaturing gradient gel electrophoresis and correspondence analysis. Infect. Genet. Evol. 2016, 44, 479–486.
- Petrova, M.I.; Lievens, E.; Malik, S.; Imholz, N.; Lebeer, S. Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol. 2015, 6, 81.
More
Information
Subjects:
Obstetrics & Gynaecology; Virology; Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
680
Revisions:
2 times
(View History)
Update Date:
30 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No