Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fabian Schramm | + 2332 word(s) | 2332 | 2022-03-21 05:16:36 | | | |
| 2 | Dean Liu | -2 word(s) | 2330 | 2022-03-30 03:01:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Schramm, F. EGFR Signaling in Lung Fibrosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/21108 (accessed on 31 July 2026).
Schramm F. EGFR Signaling in Lung Fibrosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/21108. Accessed July 31, 2026.
Schramm, Fabian. "EGFR Signaling in Lung Fibrosis" Encyclopedia, https://encyclopedia.pub/entry/21108 (accessed July 31, 2026).
Schramm, F. (2022, March 28). EGFR Signaling in Lung Fibrosis. In Encyclopedia. https://encyclopedia.pub/entry/21108
Schramm, Fabian. "EGFR Signaling in Lung Fibrosis." Encyclopedia. Web. 28 March, 2022.
Copy Citation
The epidermal growth factor (EGF) receptor (EGFR), belongs to the family of the ErbB tyrosine kinase receptors. EGFR is also known as ErbB1 or HER1. Other members of this family are ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4).
epidermal growth factor
epidermal growth factor receptor
ErbB-signaling
1. Introduction
EGF is a prototype member of the ErbB ligand family. This polypeptide was originally identified in the submaxillary glands of mice and the urine of humans. EGF was discovered by Stanley Cohen while working with Rita Levi-Montalcini on nerve growth factors. For these discoveries, Stanley Cohen and Rita Levi-Montalcini were awarded the Nobel Prize in Physiology or Medicine in 1986 [1]. EGF, TGF-α, AREG and EPG only interact with ErbB1, while EREG, BC and HB-EGF bind to ErbB1 and ErbB4. A heterogeneous group of ligands called neuregulins (NRG) interacts with ErbB3 and ErbB4 [2] (Figure 1). The NRG family is composed of NRG1 isoforms, NRG2 (also known as neural- and thymus-derived activator for ErbB kinases (NTAK)) [3][4], NRG3 [5] and NRG4 [6]. The different NRG1 isoforms can be categorized into three smaller groups: type I NRG1, which includes heregulins (HRGs) [7], neu differentiation factor (NDF) [8][9] and acetylcholine receptor-inducing activity (ARIA) [10]; type II NRG1, which contains the glial growth factors (GGFs) [8] and type III NRG1, which comprises the sensory and motor neuron-derived factor (SMDF) [11]. While NRG1 and NRG2 bind to ErbB3 and ErbB4, NRG3 and NRG4 only interact with ErbB4 [2] (see Figure 1).
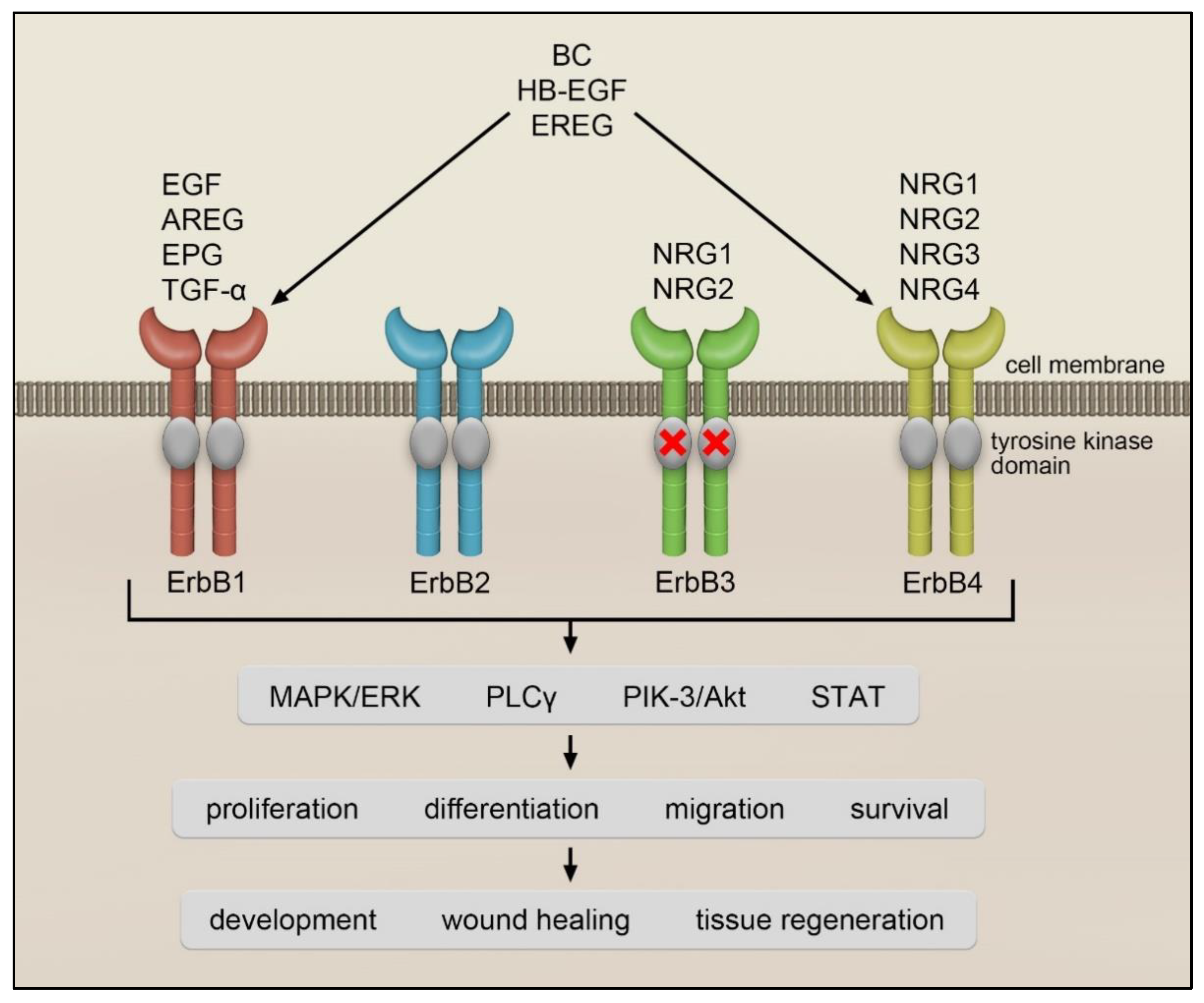
Figure 1. ErbB receptors and their ligands.
2. ErbB1/EGFR Receptor
Besides being overexpressed in many types of cancer, ErbB1 is also upregulated in lung epithelial cells from patients with different forms of pulmonary fibrosis [12]. In IPF, abundant ErbB1 immunostaining was found in the hyperplastic alveolar epithelium surrounding areas of fibrosis and inflammation. In addition, increased ErbB1 protein levels were reported in IPF fibroblastic foci and in fibroblasts isolated from IPF lungs [13]. Furthermore, IPF lung fibroblast (LF)-derived culture supernatants were found to stimulate expression of ErbB1 in donor LF in an FGF-dependent manner [13]. In addition, a negative correlation between ErbB1 mRNA levels and the indicators of IPF progression, such as forced vital capacity (FVC) and diffusion capacity of the lung for carbon monoxide (DLCO) [12], was reported.
Development of acute lung injury and ILD in non-small cell lung cancer (NSCLC) patients receiving gefitinib demonstrates possible deleterious effects of the ErbB1 signaling inhibition [14][15]. Interestingly, similar harmful effects were also observed in NSCLC patients treated with another ErbB1 TKI, erlotinib [16][17]. The incidence of ILD in TKI-treated NSCLC patients is ~1% worldwide [18]. In the Japanese population, it is significantly higher at ~2%. Despite this observation, the EGFR polymorphism leading to the genetic susceptibility to the treatment with ErbB1 TKI in the Japanese was not observed [18]. Pre-existing lung disorders, such as interstitial pneumonia or pulmonary fibrosis, male sex and history of smoking, were identified as risk factors for the development of gefitinib-associated ILD [19][20]. Considering the chemical and pharmacological similarities between gefitinib and erlotinib, the same risk factors may apply to the erlotinib-trigger ILD. In addition, radio- and chemotherapy, both used to treat cancer, seem to aggravate ErbB1 TKI-induced ILD [21]. Currently, it is not known what mechanisms lead to the development of ILD in NSCLS patients receiving gefitinib or erlotinib. However, it is increasingly recognized that the border between adaptive and maladaptive repair of the lung tissue is thin and the clue to success is maintaining the balance between all the factors involved [22].
3. Transforming Growth Factor-α
Among all ErbB1 ligands, TGF-α is the one with a well-described function in pulmonary fibrosis. TGF-α was found to be overexpressed in ATII cells, endothelial cells and fibroblasts in the lungs of IPF patients [23]. In addition, its levels were reported to be increased in IPF bronchoalveolar lavage fluid (BALF) [24]. The profibrotic potential of TGF-α was demonstrated in several studies, in which lung-specific overexpression of TGF-α in mice was conducted. For example, chronic production of TGF-α in surfactant protein-C (SP-C)-expressing cells disrupted alveolar and vascular development and caused pulmonary fibrosis and pulmonary hypertension in mice [21]. Similarly, chronic conditional expression of TGF-α driven by the doxycycline-regulatable Clara cell secretory protein (dox-CCSP) promoter triggered progressive vascular adventitial, peribronchial, interstitial, and pleural fibrosis, which was independent of inflammation and TGF-β activation [25]. Further studies demonstrated transcriptional similarities between dox-CCSP-TGF-α-induced lung fibrosis and IPF, thus pointing towards an essential role of the ErbB1–TGF-α axis in the development of IPF. In the rat bleomycin model, increased immunoreactivity for TGF-α and ErbB1 was observed in macrophages, alveolar septal cells and respiratory epithelial cells. Both proteins were predominantly detected in foci of cellular proliferation and in areas of intra-alveolar fibrosis [26]. Accordingly, TGF-α-deficient mice showed reduced hydroxyproline levels and partially preserved lung structure following bleomycin application as compared to wild-type littermates [27]. Interestingly, overexpression of TGF-α under the control of SP-C promoter protected mice against acute lung injury was caused by inhalation of polytetrafluoroethylene (PTFE; teflon) fumes. Histological hallmarks of this model are pulmonary hemorrhage and inflammation. Indeed, SP-C-TGFα-transgenic mice exhibited reduced levels of IL-6 and macrophage inflammatory protein 2 in lung homogenates and decreased total protein levels and neutrophil numbers in BALF as compared to non-transgenic controls. Altogether, these findings demonstrate the etiology-dependent role of TGF-α in lung pathologies.
In line with these observations, ErbB1 TKI, gefitinib, partially reduced collagen levels and improved lung compliance, tissue and airway elastance, and airway resistance in mice overexpressing TGF-α under tetracycline-inducible CCSP (rtTA-CCSP) promoter [25]. These changes were supported by the decreased expression of several genes associated with lung parenchymal and vascular remodeling. It is worth mentioning here that gefitinib neither induced chronic lung injury nor exacerbated pulmonary fibrosis, thus supporting further studies to determine the role of ErbB1 in human lung fibrotic diseases [21][28]. To sum up, a growing body of evidence suggests that TGF-α-driven activation of the ErbB1 signaling pathways may play an important role in the development of lung fibrosis and that TGF-α might be amenable to targeted therapy.
4. Amphiregulin
Amphiregulin, another ErbB1 ligand, was discussed in the context of maladaptive remodeling of the liver and lung. In this respect, AREG-deficient mice were found to be protected against liver fibrosis induced by chronic administration of carbon tetrachloride (CCl4) [29]. To decipher the underlying molecular mechanism, several studies focused on the link between AREG and a master regulator of fibrogenesis, TGF-β1. Zhou et al. [30] reported that stimulation of fibroblasts with TGF-β1 elevates AREG production, which in turn increases cell proliferation and the expression of profibrotic genes, such as α-smooth muscle actin, collagen 1-α1/α2, fibronectin and tenascin. These effects were reversed by the treatment of TGF-β1-stimulated fibroblasts with AREG siRNA or ErbB1 inhibitors, AG1478 or gefitinib [30]. Consistent with these in vitro findings, AREG expression was markedly increased in the lungs of dox-CC10-TGF-β1 overexpressing mice and administration of AREG siRNA or AG1478 reduced collagen content and attenuated lung fibrosis in these animals. Besides AREG, the increased expression of other ErbB1 ligands, such as EREG and HB-EGF following exposure of fibroblasts to TGF-β1, was reported [31][32]. Andrianifahanana et al. [32] documented that TGFβ-induced AREG, EREG, and HB-EGF production requires the integration of an autocrine signal from a PDGF receptor and engages a positive feedback loop through ErbB1. The same authors demonstrated the pathological relevance of PDGFR-ErbB1 cooperation in the bleomycin model of lung fibrosis. Namely, they observed that simultaneous application of imatinib (a PDGF receptor inhibitor) and lapatinib (an ErbB1/2 inhibitor) is more effective than either treatment alone. Although, there is no evidence that pirfenidone and nintedanib directly interfere with the ErbB1, their ability to inhibit TGFβ and VEGF/PDGF/FGF receptors, respectively, might influence the overall ErbB1 activity in lung fibrosis. Accordingly, Shochet et al. reported that ErbB1 expression in donor LF triggered by IPF LF-culture media depends on FGF and can be reversed by nintedanib [13]. Interconnections of ErbB1 with other signaling pathways have to be considered when designing future IPF therapies.
The complexity of AREG cellular effects is underscored by the publication by Stancil et al., who showed the role of the AREG–ErbB1 axis in the jamming–unjamming of airway epithelial cells in IPF. Jamming transition describes a process of epithelial cell transformation from migratory (unjammed) to non-migratory (jammed) status in the absence of wounding or cell-type changes. This transition is believed to play an important role during embryogenesis, in processes such as axis elongation and tissue development [33][34][35]. It was also associated with the pathogenesis of carcinomas [36] and asthma [37]. Stancil et al. [38] demonstrated in vitro that the unjammed phase is extended in distal airway epithelial cells of IPF patients and is associated with increased activity of the ErbB-YAP (Yes-Associated Protein) signaling pathway. YAP is a transcriptional co-activator, which was found to regulate epithelial progenitor cell proliferation in the lung [39] and epithelial–mesenchymal transition in lung cancer cells following exposure to TGF-β [40]. These findings are supported by the increased levels of AREG in IPF distal airway epithelial cells and its ability to induce jammed to unjammed transition in controlling distal airway epithelial cells in vitro [38]. Interestingly, the AREG-triggered extended unjammed phase of distal airway epithelial cells correlated with activation of the fibroblasts lying underneath [38], thus providing ample evidence for the contribution of airways epithelial cells repopulating distal parts of IPF lungs to the disease progression. The association between the AREG-driven prolonged unjammed status of distal airway epithelial cells and the gain-of-function MUC5B promotor variant underscores this assumption.
Besides its “bad” role in lung fibrosis, AREG was also found to contribute to the restoration of tissue homeostasis after acute lung injury driven by infection. Minutti et al. [41] showed that macrophage-derived AREG promotes TGF-β1 activation and subsequent differentiation of pericytes into collagen-producing myofibroblasts leading to restoration of vascular integrity in injured tissue and wound healing. Thus, not only TGF-β1 may regulate AREG expression but vice versa AREG can control the levels of active TGF-β1. It seems that the first scenario operates in fibrosis and the second under inflammatory conditions. Thus, the function of AREG may depend on its source, concentration and cellular and molecular landscape of the surrounding area. Although further research is needed to decipher the function of AREG in acute versus chronic pathological conditions, it becomes clear that identification of dynamics and causal flows in complex AREG signaling networks is crucial for its use as a therapeutic target. This assumption is supported by the study demonstrating attenuation of bleomycin-induced lung fibrosis upon AREG application during the late inflammatory phase [42]. This observation is in sharp contrast to the lung fibrosis reports mentioned above but it may be explained by the findings of Minutti et al. [41], namely, the AREG effects on blood vessel regeneration and thus epithelial cell survival in acute lung injury [30]. Overall, it seems that AREG properties may depend on the genetic background and the immune system condition, thus preselecting the potential responders prior to the treatment may raise the possibility of the success of an anti-AREG therapy in IPF.
5. ErbB2 and ErbB3 Receptors and Their Ligands
Another ErbB receptor that was linked to pulmonary fibrosis is ErbB2. Besides being an important oncogene in breast and ovarian cancer [43], ErbB2 was found to be involved in epithelial cell recovery upon acute lung injury. While ErbB2 was detected on the basolateral side of airway epithelial cells, HRG-α was only found in the apical membrane of these cells and in the overlying mucus film [44]. When epithelial integrity is disrupted, HRG-α translocates to Erb2 and enables a rapid response to injury. Thus, the Erb2-HRG-α systems sense changes in the extracellular environment and ensure restoration of barrier function that may be critical for survival. As there is no known ligand for ErbB2, this receptor is able to transduce intracellular signaling only upon forming a complex with other ErbB receptors. In pulmonary epithelial cells, ErbB2 is the preferred binding partner for ErbB3. Besides being engaged in the HRG-α-triggered intracellular signaling, the ErbB2–ErbB3 complex also responds to NRG1. Using a dominant-negative mutant of ErbB3 expressed under the SP-C promotor, Nethery et al. [45] demonstrated that SP-C-ErbB3 transgenic mice exhibit reduce collagen levels in the lung and better survival following bleomycin administration. The effect was associated with the inability of NRG1 to signal via the nonfunctional ErbB2–ErbB3 complex. These findings are corroborated by Faress et al. [46], who reported preserved lung structure and diminished lung collagen content upon administration of an anti-ErbB2 antibody (2C4) to bleomycin-treated mice.
The important role of ErbB2 in bronchial epithelial cell differentiation and proliferation was shown by Vermeer et al. [47]. These authors demonstrated that treatment of airway epithelial cells with an anti-ErbB2 antibody, trastuzumab, induces their de-differentiation associated with an increase in the numbers of non-ciliated and metaplastic, flat cells. By contrast, the exposure of the cells to HRG-α preserved normal differentiation of airway epithelial cells. Most interestingly, co-culturing of airway epithelial cells with fibroblasts potentiated epithelial cell differentiation comparable to that achieved following treatment with HRG-α, pointing towards the ability of fibroblasts to produce ErbB ligands. Indeed, further studies demonstrated that normal human LF express TGF-α, HB-EGF, EREG, AREG, and HRG-α [47]. These observations were in line with the clinical case report describing reversible changes in airway epithelial cell differentiation of a breast cancer patient that coincided with the initiation and discontinuation of a trastuzumab therapy [47].
The ErbB3–NRG1-α axis was also discussed in the context of alveolar bronchiolization seen in the lungs of IPF patients. In these patients, NRG1-α was detected in epithelial cells lining honeycombing areas, as well as in normal submucosal glands [48]. In addition, elevated levels of this molecule were measured in IPF BALF [49]. Given the ability of NRG1-α to regulate airway mucus cell differentiation and MUC5B expression, it is worth speculating about its pivotal role in airway epithelial cell reprogramming and thus honeycomb cyst formation in IPF [50].
Taken together, it seems that ErbB2–ErbB3 activation is essential for the differentiation of airway epithelial cells and their integrity. Overactivation of this receptor complex system may induce abnormal behavior of airway epithelial cells thereby contributing to the honeycomb cyst formation and fibrosis progression. Because of the risks associated with the ErbB2–ErbB3 complex inhibition, close patient monitoring and patient categorization have to be taken into account when considering an anti-ErbB2–ErbB3 therapy in IPF.
References
- NobelPrize.org. Available online: https://www.nobelprize.org/prizes/medicine/1986/summary/ (accessed on 22 January 2022).
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nature reviews. Mol. Cell Biol. 2001, 2, 127–137.
- Higashiyama, S.; Abraham, J.A.; Miller, J.; Fiddes, J.C.; Klagsbrun, M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 1991, 251, 936–939.
- Chang, H.; Riese, D.J.; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512.
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567.
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 1999, 18, 2681–2689.
- Holmes, W.E.; Sliwkowski, M.X.; Akita, R.W.; Henzel, W.J.; Lee, J.; Park, J.W.; Yansura, D.; Abadi, N.; Raab, H.; Lewis, G.D. Identification of heregulin, a specific activator of p185erbB2. Science 1992, 256, 1205–1210.
- Wen, D.; Peles, E.; Cupples, R.; Suggs, S.V.; Bacus, S.S.; Luo, Y.; Trail, G.; Hu, S.; Silbiger, S.M.; Levy, R.B.; et al. Neu differentiation factor: A transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992, 69, 559–572.
- Peles, E.; Bacus, S.S.; Koski, R.A.; Lu, H.S.; Wen, D.; Ogden, S.G.; Levy, R.B.; Yarden, Y. Isolation of the NeuHER-2 stimulatory ligand: A 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell 1992, 69, 205–216.
- Falls, D. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell 1993, 72, 801–813.
- Ho, W.H.; Armanini, M.P.; Nuijens, A.; Phillips, H.S.; Osheroff, P.L. Sensory and motor neuron-derived factor. A novel heregulin variant highly expressed in sensory and motor neurons. J. Biol. Chem. 1995, 270, 14523–14532.
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. BioMed Res. Int. 2013, 2013, 654354.
- Epstein Shochet, G.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L1025–L1034.
- Inomata, S.; Takahashi, H.; Nagata, M.; Yamada, G.; Shiratori, M.; Tanaka, H.; Satoh, M.; Saitoh, T.; Sato, T.; Abe, S. Acute lung injury as an adverse event of gefitinib. Anti-Cancer Drugs 2004, 15, 461–467.
- Inoue, A.; Saijo, Y.; Maemondo, M.; Gomi, K.; Tokue, Y.; Kimura, Y.; Ebina, M.; Kikuchi, T.; Moriya, T.; Nukiwa, T. Severe acute interstitial pneumonia and gefitinib. Lancet 2003, 361, 137–139.
- Ten Heine, R.; van den Bosch, R.T.A.; Schaefer-Prokop, C.M.; Lankheet, N.A.G.; Beijnen, J.H.; Staaks, G.H.A.; van der Westerlaken, M.M.; Malingré, M.M.; van den Brand, J.J.G. Fatal interstitial lung disease associated with high erlotinib and metabolite levels. A case report and a review of the literature. Lung Cancer 2012, 75, 391–397.
- Ren, S.; Li, Y.; Li, W.; Zhao, Z.; Jin, C.; Zhang, D. Fatal asymmetric interstitial lung disease after erlotinib for lung cancer. Respir. Int. Rev. Thorac. Dis. 2012, 84, 431–435.
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa) tablets. Oncologist 2003, 8, 303–306.
- Akamatsu, H.; Inoue, A.; Mitsudomi, T.; Kobayashi, K.; Nakagawa, K.; Mori, K.; Nukiwa, T.; Nakanishi, Y.; Yamamoto, N. Interstitial lung disease associated with gefitinib in Japanese patients with EGFR-mutated non-small-cell lung cancer: Combined analysis of two Phase III trials (NEJ 002 and WJTOG 3405). Jpn. J. Clin. Oncol. 2013, 43, 664–668.
- Ando, M.; Okamoto, I.; Yamamoto, N.; Takeda, K.; Tamura, K.; Seto, T.; Ariyoshi, Y.; Fukuoka, M. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2549–2556.
- Hardie, W.D.; Davidson, C.; Ikegami, M.; Leikauf, G.D.; Le Cras, T.D.; Prestridge, A.; Whitsett, J.A.; Korfhagen, T.R. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 294, L1217–L1725.
- Li, C.; Wei, R.; Jones-Hall, Y.L.; Vittal, R.; Zhang, M.; Liu, W. Epidermal growth factor receptor (EGFR) pathway genes and interstitial lung disease: An association study. Sci. Rep. 2014, 4, 4893.
- Baughman, R.P.; Lower, E.E.; Miller, M.A.; Bejarano, P.A.; Heffelfinger, S.C. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 1999, 16, 57–61.
- Madtes, D.K.; Rubenfeld, G.; Klima, L.D.; Milberg, J.A.; Steinberg, K.P.; Martin, T.R.; Raghu, G.; Hudson, L.D.; Clark, J.G. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1998, 158, 424–430.
- Hardie, W.D.; Le Cras, T.D.; Jiang, K.; Tichelaar, J.W.; Azhar, M.; Korfhagen, T.R. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L741–L749.
- Madtes, D.K.; Busby, H.K.; Strandjord, T.P.; Clark, J.G. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am. J. Respir. Cell Mol. Biol. 1994, 11, 540–551.
- Madtes, D.K.; Elston, A.L.; Hackman, R.C.; Dunn, A.R.; Clark, J.G. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am. J. Respir. Cell Mol. Biol. 1999, 20, 924–934.
- Hardie, W.D.; Korfhagen, T.R.; Sartor, M.A.; Prestridge, A.; Medvedovic, M.; Le Cras, T.D.; Ikegami, M.; Wesselkamper, S.C.; Davidson, C.; Dietsch, M.; et al. Genomic profile of matrix and vasculature remodeling in TGF-alpha induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 309–321.
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goñi, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261.
- Zhou, Y.; Lee, J.-Y.; Lee, C.-M.; Cho, W.-K.; Kang, M.-J.; Koff, J.L.; Yoon, P.-O.; Chae, J.; Park, H.-O.; Elias, J.A.; et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000.
- Andrianifahanana, M.; Wilkes, M.C.; Repellin, C.E.; Edens, M.; Kottom, T.J.; Rahimi, R.A.; Leof, E.B. ERBB receptor activation is required for profibrotic responses to transforming growth factor beta. Cancer Res. 2010, 70, 7421–7430.
- Andrianifahanana, M.; Wilkes, M.C.; Gupta, S.K.; Rahimi, R.A.; Repellin, C.E.; Edens, M.; Wittenberger, J.; Yin, X.; Maidl, E.; Becker, J.; et al. Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4444–4454.
- Mongera, A.; Rowghanian, P.; Gustafson, H.J.; Shelton, E.; Kealhofer, D.A.; Carn, E.K.; Serwane, F.; Lucio, A.A.; Giammona, J.; Campàs, O. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature 2018, 561, 401–405.
- Atia, L.; Fredberg, J.J.; Gov, N.S.; Pegoraro, A.F. Are cell jamming and unjamming essential in tissue development? Cells Dev. 2021, 4, 203727.
- Bi, D.; Lopez, J.H.; Schwarz, J.M.; Manning, M.L. A density-independent glass transition in biological tissues. Nat. Phys. 2015, 11, 1074–1079.
- Palamidessi, A.; Malinverno, C.; Frittoli, E.; Corallino, S.; Barbieri, E.; Sigismund, S.; Beznoussenko, G.V.; Martini, E.; Garre, M.; Ferrara, I.; et al. Unjamming overcomes kinetic and proliferation arrest in terminally differentiated cells and promotes collective motility of carcinoma. Nat. Mater. 2019, 18, 1252–1263.
- Park, J.-A.; Atia, L.; Mitchel, J.A.; Fredberg, J.J.; Butler, J.P. Collective migration and cell jamming in asthma, cancer and development. J. Cell Sci. 2016, 129, 3375–3383.
- Stancil, I.T.; Michalski, J.E.; Davis-Hall, D.; Chu, H.W.; Park, J.-A.; Magin, C.M.; Yang, I.V.; Smith, B.J.; Dobrinskikh, E.; Schwartz, D.A. Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat. Commun. 2021, 12, 4566.
- Lange, A.W.; Sridharan, A.; Xu, Y.; Stripp, B.R.; Perl, A.-K.; Whitsett, J.A. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol. 2015, 7, 35–47.
- Choi, J.-Y.; Lee, H.; Kwon, E.-J.; Kong, H.-J.; Kwon, O.-S.; Cha, H.-J. TGFβ promotes YAP-dependent AXL induction in mesenchymal-type lung cancer cells. Mol. Oncol. 2021, 15, 679–696.
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654.e6.
- Fukumoto, J.; Harada, C.; Kawaguchi, T.; Suetsugu, S.; Maeyama, T.; Inoshima, I.; Hamada, N.; Kuwano, K.; Nakanishi, Y. Amphiregulin attenuates bleomycin-induced pneumopathy in mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2010, 298, L131–L138.
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712.
- Vermeer, P.D.; Einwalter, L.A.; Moninger, T.O.; Rokhlina, T.; Kern, J.A.; Zabner, J.; Welsh, M.J. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 2003, 422, 322–326.
- Nethery, D.E.; Moore, B.B.; Minowada, G.; Carroll, J.; Faress, J.A.; Kern, J.A. Expression of mutant human epidermal receptor 3 attenuates lung fibrosis and improves survival in mice. J. Appl. Physiol. 2005, 99, 298–307.
- Faress, J.A.; Nethery, D.E.; Kern, E.F.O.; Eisenberg, R.; Jacono, F.J.; Allen, C.L.; Kern, J.A. Bleomycin-induced pulmonary fibrosis is attenuated by a monoclonal antibody targeting HER2. J. Appl. Physiol. 2007, 103, 2077–2083.
- Vermeer, P.D.; Panko, L.; Karp, P.; Lee, J.H.; Zabner, J. Differentiation of human airway epithelia is dependent on erbB2. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L175–L180.
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657.
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775.
- Kettle, R.; Simmons, J.; Schindler, F.; Jones, P.; Dicker, T.; Dubois, G.; Giddings, J.; van Heeke, G.; Jones, C.E. Regulation of neuregulin 1beta1-induced MUC5AC and MUC5B expression in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2010, 42, 472–481.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
2 times
(View History)
Update Date:
30 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No