Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Konstantinos Gousias | + 2435 word(s) | 2435 | 2022-03-11 04:51:18 | | | |
| 2 | Amina Yu | -6 word(s) | 2429 | 2022-03-28 04:47:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gousias, K. Cell Cycle Arrest and Apoptosis in Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/21101 (accessed on 29 June 2026).
Gousias K. Cell Cycle Arrest and Apoptosis in Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/21101. Accessed June 29, 2026.
Gousias, Konstantinos. "Cell Cycle Arrest and Apoptosis in Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/21101 (accessed June 29, 2026).
Gousias, K. (2022, March 28). Cell Cycle Arrest and Apoptosis in Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/21101
Gousias, Konstantinos. "Cell Cycle Arrest and Apoptosis in Glioblastoma." Encyclopedia. Web. 28 March, 2022.
Copy Citation
Glioblastoma multiforme (GBM) account for 49% of all primary malignant central nervous system tumorsand stand for the most malignant part of the clinical spectrum. The current established therapy consists of a gross total resection when safely feasible, followed by adjuvant radio-, chemo-, or radiochemotherapy and application of tumor treating fields (TTF). Nevertheless, prognosis is still poor, and overall survival after completion of these therapies averages less than 2 years.
glioblastoma
cell cycle arrest
apoptosis
p53 pathway
Rb pathway
ion channels
1. Aberrant Cell Cycle Progression and Apoptosis in Glioblastoma multiforme (GBM)
Dysregulation of a variety of cellular pathways, in particular those involved in the regulation of the cell cycle machinery and apoptosis, is observed in several types of cancer. Subsequently, tumor cells may escape apoptosis or senescence and show excessive proliferation and tumor growth. One can argue that apoptosis and senescence protect against cancer. The most common alterations found in GBM affect p53 [1]; 30% of primary and 65% of secondary GBM express mutated p53 [2]. Both missense and splice site mutations are observed [3]; in the case of the former, several hotspots in the DNA-binding domain—namely, R175, R248, R249, R273, R273, R282, and G245—are most frequently mutated, according to the GBM PanCancer Atlas of The Cancer Genome Atlas (TCGA) [4][5][6]. In addition, methylation of the p53 gene promoter was detected in 21% of primary GBM in one study [7]. Apart from loss or mutation of the p53 gene, further mechanisms that result in p53 inactivation in GBM include impairment of p53 protein stability and suppression of p53 gene expression through amplification of p53 inhibitor genes, such as MDM2 and MDM4 [8][9], genetic deletion and methylation of the p53 inducer ARF [10], genomic loss of ATM, CHEK2 [11], mutation of Parkin [12], overexpression of NFIA, and miR-141-3o [13][14], Bcl2 [15][16], and MIF [17].
According to the TCGA data, prominent alterations in the Rb pathway include homozygous deletions or mutations of genes coding for members of the pocket protein family, in particular of Rb, and gene amplification of cell cycle promoters such as CDKs (CDK4 and 6) and cyclin D1 [18]. Mutation, deletion, or methylation of Rb is observed more frequently in secondary GBM [19].
The P13K/AKT/mTOR pathway is upregulated in GBM cell lines, such as U138 MG [20]. Homozygous deletions or mutations of PTEN, mutations of P13K, as well as amplification of AKT and FOXO genes are documented in the TCGA [18]. PTEN mutations have been associated with poor survival in GBM patients [21].
A higher expression of p38 showed a positive correlation with the WHO grade of malignancy in gliomas, implying also an aberrant activity of the MAPK pathway [22]. Stem cell GBM cells showed self-renewing ability upon phosphorylation of JNK; the systemic administration of small-molecule JNK inhibitors blocks this ability [23].
The NF-kB pathway, which demonstrates anti-apoptotic activity, is upregulated in GBM cells. The NF-kB p65 subunit is overexpressed in gliomas, showing a positive correlation with the WHO malignancy [24]. Inhibition of the NF-kB subunits RelA and c-Rel drives cell cycle arrest and reduction in tumor growth in GBM cells [25]. Significant interactions between NF-kB and p53 cascades in GBM promote cell cycle arrest, apoptosis, neovascularization, impaired EGFR signaling, and neuroinflammation [26] (Figure 1).
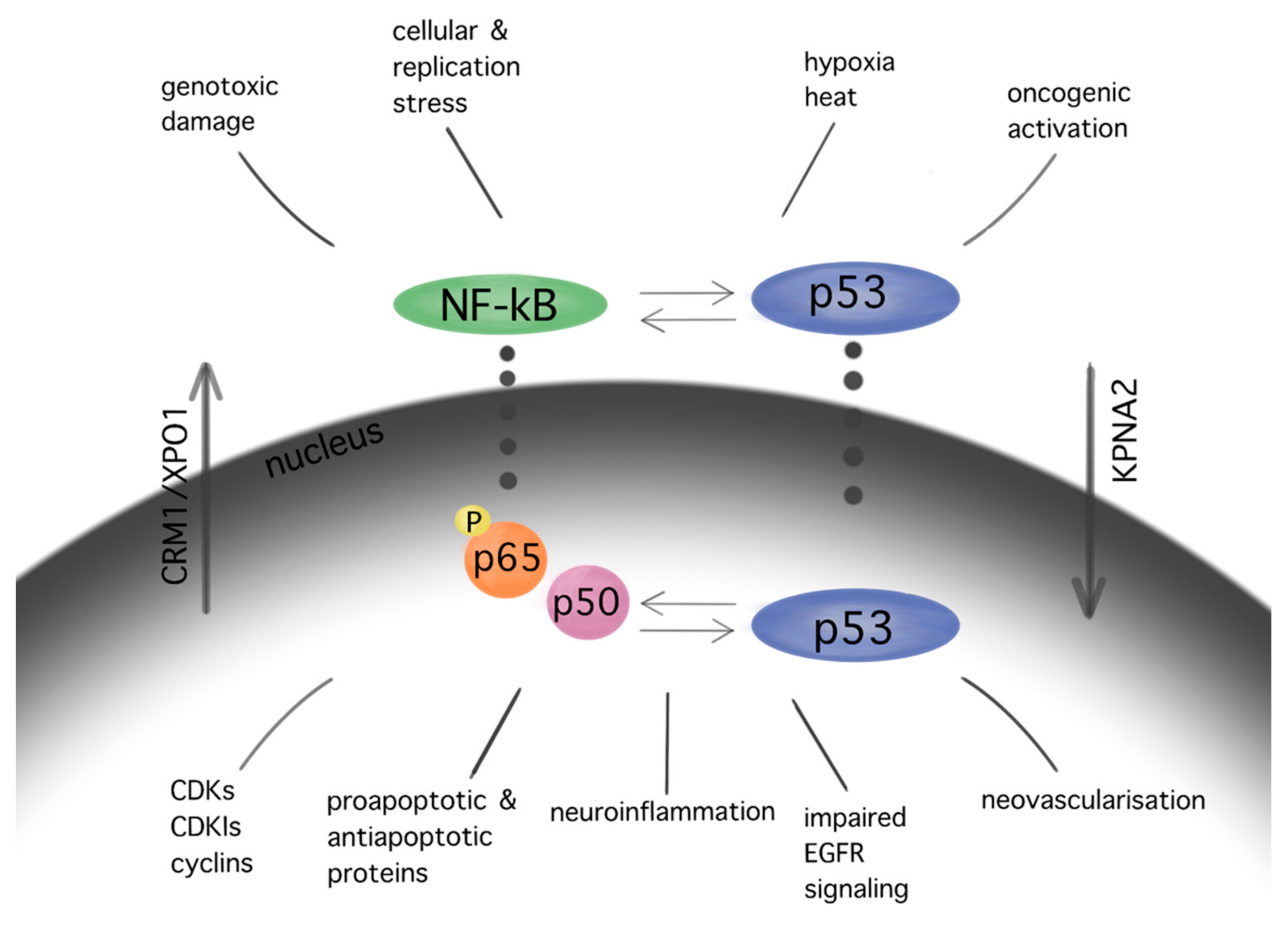
Figure 1. Interactions between pathways of p53 and NF-kB in GBM. Similar stimuli trigger NF-kB and p53 pathways. In turn, several interactions between the aforementioned cascades are observed in multiple levels at their cytoplasmic as well as their nuclear localization, resulting in cell cycle arrest, apoptosis, neuroinflammation, impaired EGFR signaling, and angiogenesis in GBM. The subcellular translocation is performed by karyopherins (nuclear import: KPNA2; nuclear export: CRM1/XPO1).
Mutations in the TERT gene promoter underlie a further escape mechanism from apoptosis in GBM; GBM cells maintain the telomere length in the context of increased telomerase activity, and this in turn leads to excessive proliferation. TERT promoter mutations are frequently observed in IDH-wildtype GBM [27][28]. TERT promoter mutations have been correlated with shorter survival [29].
In vitro and in vivo studies showed an aberrant nucleocytoplasmic transport in patients with GBM or GBM cell lines [30][31]. KPNA2 and CRM1 are upregulated in brain tumors, whereas their expression correlated positively with the WHO malignancy grade [32][31]. KPNA2 expression, in particular, showed an inverse correlation with the patients’ overall and progression-free survival. Increased KPNA2 expression in the UM87 GBM cell line was associated with more malignant behavior via activation of the p53 pathway [30].
2. Principles of GBM Molecular Targeting
2.1. Current Therapy in GBM and Cell Cycle Control
The current therapy of patients with GBM consists of maximal safe tumor resection followed by adjuvant radiation and chemotherapy. Different types of radiation and chemotherapy are used for newly diagnosed vs. recurrent GBM and for disparate tumor biology. Additional application of alternating electrical (“tumor treating”, TTF) fields appear as a safe and promising additional therapy [33].
Radiation induces a variety of DNA lesions, such as damaged bases and DNA strand breaks. Approximately 1000 single and 40 double strand breaks are produced per Gy per cell [34]. The radiation-induced DNA damage is monitored by the kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related protein (ATR), which in turn initiate the DNA damage response, as previously described [35]. As a consequence, the tumor cells initially undergo cell cycle arrest, and in case of a serious unrepairable DNA damage, cell death via mitotic catastrophe and apoptosis [34]. The effect of radiation therapy depends on the total dose, the number of fractions applied [36], as well as the quality of radiation [37]. The initiation of the mitotic catastrophe occurs not immediately after radiation but rather after accumulation of sufficient genetic damage, reflecting the delayed clinical and imaging response of GBM to radiation.
Combined treatment with temozolomide and “tumor treating” (TTF) prolonged the overall and progression-free survival of patients with newly diagnosed GBM (EF-14 trial) [33]. TTF force dipole alignment and dielectrophoresis of proteins involved in spindle formation and mitosis, such as septin 2, 6, and 7, a/b tubulin, and microtubules of spindles [38]. The impaired formation of microtubules induces cytoplasmic blebbing, mitotic failure, and abnormal chromosome segregation, with subsequent disruption of mitosis and cell death via apoptosis [39].
The cytotoxicity of the alkylating agent temozolomide is mediated among others by the addition of methyl groups at O6 sites on guanines in genomic DNA, which in turn causes base mispairing [40]. In more detail, their toxic product O6-methylguanine is then paired with thymine instead of cytosine during DNA replication. The mismatched O6-methylguanine to thymine base pair is sensed by DNA repair pathways involving the repair proteins MLH1, MSH2, MSH6, and PMS2, which place the tumor cells initially into cell cycle arrest and eventually cause cell death. However, approximately 60% of patients with GBM show resistance to temozolomide, since a nuclear enzyme, named O6-methylguanine-DNA methyltransferase (MGMT), removes alkyl groups from the O6-position of O6-methylguanine and returns the cell into the regular cell cycle mode. The methylation status of the MGMT promoter, which silences MGMT expression, has been identified as being a beneficial prognostic predictor in patients undergoing TMZ chemotherapy [41].
Nitrosoureas are anticancer agents used in the therapy of recurrent GBM but also for newly diagnosed GBM with MGMT promoter hypermethylation [42][43]. Lomustine or CCNU, a well-known nitrosourea, transfers its chloroethyl group to the O6 sites of guanine on DNA. This causes interstrand and intrastrand cross-linking of DNA, which inactivates DNA synthesis and leads to cell death. Similar to temozolomide, O6-methylguanine DNA methyltransferase (MGMT) also reverts the product of CCNU—namely, the of O6-chloroethylguanine, removing its alkyl group, restricting the meaningful use of CCNU in patients with methylated MGMT [44].
2.2. Targeting the Cell Cycle Machinery in GBM
Since GBM cells show uncontrolled cell cycle progression due to alterations of the p53 and Rb pathway, many studies have focused on restoring these functions [45]. Mutations of p53 and Rb are the most common sources of impairments, but direct targeting of p53 and Rb mutations is challenging. However, alternative ways of pathways’ modulations, such as inhibition of natural p53 and Rb deactivators, such as MDM2 or CDKIs, may be both feasible and promising [46][47][48][49][50][51]. Nutlin-3 is a MDM2 inhibitor that targets the MDM2–p53 interaction, inhibiting GBM cell growth via upregulation of apoptosis and senescence [49]. A second generation nutlin analogue called RG7388 is currently under evaluation in conjunction with radiation in the context of the NOA-20 trial (NCT03158389, https://clinicaltrials.gov/ct2/show/NCT03158389, accessed on 30 December 2021). Piperidinones, such as AMG232, are further MDM2–p53 interaction inhibitors, which are tested in a phase I clinical trial in primary and recurrent GBM (NCT03107780) (https://clinicaltrials.gov/ct2/show/NCT03107780, accessed on 30 December 2021). Alternative ways of restoring p53 functions are direct blocking of MDM2 expression via siRNA [52] or restoration of p53 expression via a p53 activator, such as RITA [53]. Similarly, targeting the Rb pathway and CDKs or cyclins drives GBM cells to cell cycle arrest in GBM models [54][55]. CDK4 and CDK6 inhibitors, which showed promising activity in various type of cancers [56], are currently under evaluation in the NCT02345824 ongoing GBM trial (https://clinicaltrials.gov/ct2/show/NCT02345824, accessed on 30 December 2021). TG02, a novel CDK9 inhibitor, is being studied in the clinical trials NCT02942264 and NCT03224104 for recurrent and newly diagnosed GBM, respectively (https://clinicaltrials.gov/ct2/show/NCT02942264; https://clinicaltrials.gov/ct2/show/NCT03224104, accessed on 30 December 2021) [57].
A variety of natural substances have been identified as being physiological regulators of the cell cycle via p53 and Rb [58]. Compared with synthetic anticancer agents, they demonstrate a diminished drug toxicity and higher permeability through the BBB [58]. The family of natural compounds comprise among others plant derivatives, curcuminoids, coumarins, alkaloids, carotenoids, flavonoids, marine peptides, and natural steroids [59][60]. The biochemical structures of the studied natural compounds but also the remaining therapeutic agents mentioned are shown in the Table 1. Curcumin upregulates CDKN2A/p16 in DBTRG glial cells, which in turn inhibits phosphorylation of Rb, which leads to a G1/S cell cycle arrest [61]. An increased BAX/BCL2 ratio is also caused by curcumin, inducing apoptosis in a p53-dependent manner via intrinsic mitochondrial pathways [62]. In addition, curcumin is reported to modulate the JAK/STAT, MAPK, p13k/Akt, and NF-kB pathways in favor of cell cycle arrest [60]. Flavonoids such as alkylaminophenol [63] and tectorigenin [64] are metabolites of plants, which promote a p53-, Rb-, and CDK-mediated cell cycle arrest and apoptosis [64][63]. An additional plant compound, named moschamine, activates the intrinsic pathway of apoptosis via dysregulation of the mitochondrial membrane potential, whereas the combined exposure of GBM cell lines to moschamine and temozolomide promotes a stronger cell cycle arrest compared with sole temozolomide exposure [65].
Table 1. Biochemical structures of potential therapeutic agents in GBM.
| Therapeutic Agents | Biochemical Structure |
|---|---|
| Nutlin-3 | 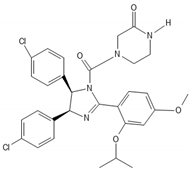 |
| RITA | 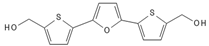 |
| Ribociclib | 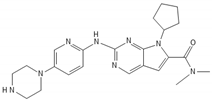 |
| TG02 |  |
| Curcumin |  |
| Moschamine |  |
| Flavonoids |  |
| JQ1 | 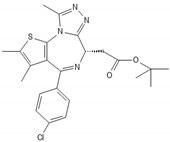 |
| UM-002 |  |
| Thiabenzole | 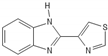 |
| Flubendazole | 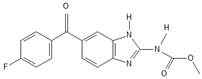 |
| 5-ALA |  |
| Haloperidol | 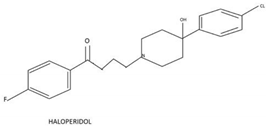 |
| Selinexor |  |
The bromodomain and extraterminal (BET) family proteins are epigenetic regulators of gene transcription by binding via their two tandem bromodomains to lysine-acetylated histones. Since BET proteins regulate the transcription of specific oncogenes as well as cell cycle related genes [66][67][68] they have been investigated as potential therapeutic targets in various cancers [69]. BET inhibitors such as JQ1 [70] induce apoptosis in glioma stem cells by modulating P13K/AKT. A novel BET inhibitor, UM-002, reduced the cell proliferation in patient-derived xenograft GBM cell lines GBM22 and GBM39 [68]. MicroRNAs are non-coding RNAs that regulate gene expression of cell cycle regulatory pathways [71]. Downregulation of microRNA-21 induces in GBM cell lines a G0/G1 cell cycle arrest and increased apoptosis and inhibits chemotherapeutic resistance to doxorubicin [72].
Reassigning a novel role to already established drugs known to be safe is a potentially promising concept in medical oncology. The benzimidazole carbamate family compounds were initially used for the treatment of anthelminthics, but they have shown additional anticancer behavior [73][74]. Hu et al. analyzed the effect of thiabenzole on GBM cell lines (P3, U251, LN229, A172, and U118MG). Thiabenzole was found to induce a G2/M arrest in GBM cell lines via downregulation of mini-chromosome maintenance protein 2 [75]. Flubendazole induces apoptosis via increasing the expression of proapoptotic proteins; in addition, cell cycle arrest is being promoted through downregulation of cyclin B1 and upregulation of p53 and CDKIs, such as p21 in GBM cells [76][77]. Recently, antipsychotic drugs have emerged as potential anticancer agents, whereas 12 candidate substances have been identified [78][79]. Treatment with haloperidol, in particular, has been reported to promote G2/M cell cycle arrest in the U87 GBM cell line [80].
5-Aminolevulinic acid (5-ALA), which induces accumulation of the protoporphyrin IX in GBM cells, is well known as the main diagnostic agent that differentiates the tumor-infiltrated tissue from adjacent healthy brain parenchyma during fluorescence-guided brain surgery [81]. Recently 5-ALA has been assigned a new role either in the context of photodynamic therapy [82] or as a direct cytotoxic agent for GBM [83]. Jalili-Nik et al. report a reduction in Bcl-2 and an increase in Bax and p53 expression, and therefore an increase in apoptotic cells, in the U87MG GBM cell line after in vitro application of 5-ALA [83].
Trans- and intracellular shuttling is a fundamental process enabling crucial cell functions, such as the regulation of cell cycle [30][84]. Transmembrane ion channels regulate the responses of the cells to external stimuli, whereas karyopherins translocate macromolecules through the nuclear envelope. Various ion channels, such as Kv10.1, NaV1.6, VDAC2, or CLIC1 are dysregulated in GBM [85][86][72]; higher expressions of TRM3, P2RX4, or CLIC1 are linked to poorer survival [86][72][87]. Since dysregulated ion channels drive tumorigenesis and cell proliferation of GBM cells, their inhibition leads to senescence or apoptosis. Inhibition of the ether-a-go-go-related gene encodes the pore-subunit of K+ channel Kv11.1 via siRNA-mediated apoptosis in GBM cell lines [88]. In vitro suppression of the Ca2+-activated K+ channel BK via its inhibitor, called iberiotoxin, induced S phase arrest and apoptosis [89].
Karyopherins are essential in cell cycle control, as they translocate relevant transcription factors, such as E2F1 and tumor suppressors, as well as oncogenes, through the nuclear envelope [90]. SiRNA-mediated silencing of the most well-characterized importin, karyopherin a2, in U87MG GBM cell line was found to induce cell cycle arrest and apoptosis in a p53-dependent manner [30]. Inhibition of the importin XPO1 or CRM1 via selinexor has reduced proliferation and prolonged survival in GBM animal models [91]. A phase 2 study on efficacy, safety, and intratumoral pharmacokinetics of selinexor monotherapy in recurrent GBM (KING Trial) [92] concluded that there was a clinically relevant response in patients with GBM to a 80 mg weekly dose of selinexor in terms of prolonged progression-free survival [92]. The follow-up study NCT04421378 analyses the effect of selinexor in combination with standard of care therapy for newly diagnosed or recurrent GBM (https://clinicaltrials.gov/ct2/show/NCT04421378, accessed on 30 December 2021). A graphic presentation of the target points of the aforementioned therapeutic agents is given in Figure 2.
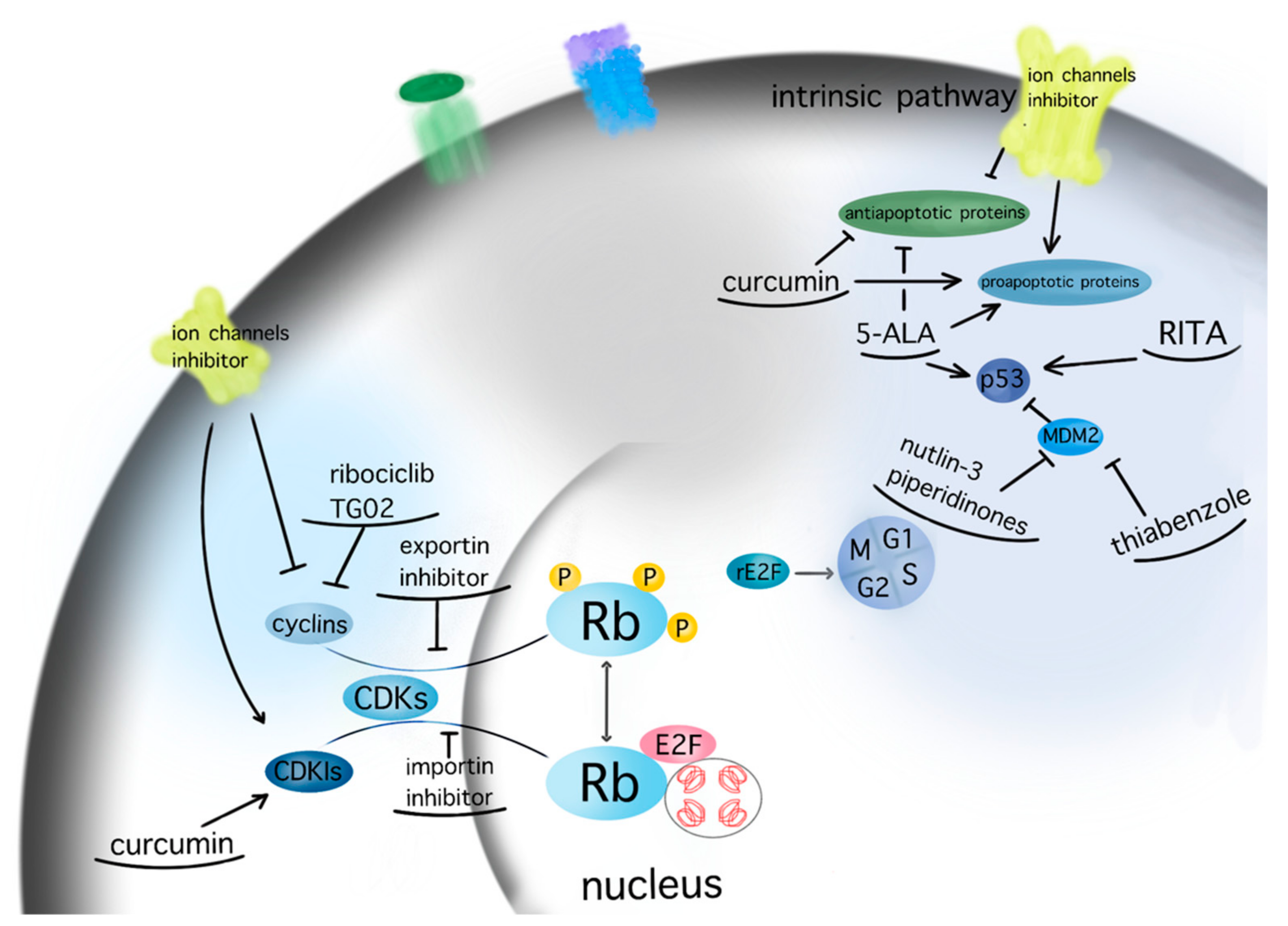
Figure 2. Molecular targeting of GBM therapeutic agents. The current figure depicts the molecular targets of potential therapeutic agents for GBM within the pathways of apoptosis and cell cycle (arrow: upregulation; line: downregulation).
References
- Hill, J.R.; Kuriyama, N.; Kuriyama, H.; Israel, M.A. Molecular genetics of brain tumors. Arch. Neurol. 1999, 56, 439–441.
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297.
- Xiong, Y.; Zhang, Y.; Xiong, S.; Williams-Villalobo, A.E. A Glance of p53 Functions in Brain Development, Neural Stem Cells, and Brain Cancer. Biology 2020, 9, 285.
- Uno, M.; Oba-Shinjo, S.M.; de Aguiar, P.H.; Leite, C.C.; Rosemberg, S.; Miura, F.K.; Junior, R.M.; Scaff, M.; Nagahashi Marie, S.K. Detection of somatic TP53 splice site mutations in diffuse astrocytomas. Cancer Lett. 2005, 224, 321–327.
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703.
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020, 79, 58–67.
- Jesionek-Kupnicka, D.; Szybka, M.; Malachowska, B.; Fendler, W.; Potemski, P.; Piaskowski, S.; Jaskolski, D.; Papierz, W.; Skowronski, W.; Och, W.; et al. TP53 promoter methylation in primary glioblastoma: Relationship with TP53 mRNA and protein expression and mutation status. DNA Cell Biol. 2014, 33, 217–226.
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739.
- Riemenschneider, M.J.; Buschges, R.; Wolter, M.; Reifenberger, J.; Bostrom, J.; Kraus, J.A.; Schlegel, U.; Reifenberger, G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999, 59, 6091–6096.
- Nakamura, M.; Watanabe, T.; Klangby, U.; Asker, C.; Wiman, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol. 2001, 11, 159–168.
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629.
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775.
- Lee, J.S.; Xiao, J.; Patel, P.; Schade, J.; Wang, J.; Deneen, B.; Erdreich-Epstein, A.; Song, H.R. A novel tumor-promoting role for nuclear factor IA in glioblastomas is mediated through negative regulation of p53, p21, and PAI1. Neuro-Oncology 2014, 16, 191–203.
- Zhou, X.; Wu, W.; Zeng, A.; Nie, E.; Jin, X.; Yu, T.; Zhi, T.; Jiang, K.; Wang, Y.; Zhang, J.; et al. MicroRNA-141-3p promotes glioma cell growth and temozolomide resistance by directly targeting p53. Oncotarget 2017, 8, 71080–71094.
- Stegh, A.H.; Brennan, C.; Mahoney, J.A.; Forloney, K.L.; Jenq, H.T.; Luciano, J.P.; Protopopov, A.; Chin, L.; Depinho, R.A. Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes Dev. 2010, 24, 2194–2204.
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111.
- Fukaya, R.; Ohta, S.; Yaguchi, T.; Matsuzaki, Y.; Sugihara, E.; Okano, H.; Saya, H.; Kawakami, Y.; Kawase, T.; Yoshida, K.; et al. MIF Maintains the Tumorigenic Capacity of Brain Tumor-Initiating Cells by Directly Inhibiting p53. Cancer Res. 2016, 76, 2813–2823.
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Grzmil, M.; Hemmings, B.A. Deregulated signalling networks in human brain tumours. Biochim. Biophys. Acta 2010, 1804, 476–483.
- Zanotto-Filho, A.; Braganhol, E.; Edelweiss, M.I.; Behr, G.A.; Zanin, R.; Schroder, R.; Simoes-Pires, A.; Battastini, A.M.; Moreira, J.C. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. J. Nutr. Biochem. 2012, 23, 591–601.
- Koul, D. PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325.
- Yang, K.; Liu, Y.; Liu, Z.; Liu, J.; Liu, X.; Chen, X.; Li, C.; Zeng, Y. p38gamma overexpression in gliomas and its role in proliferation and apoptosis. Sci. Rep. 2013, 3, 2089.
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516.
- Raychaudhuri, B.; Han, Y.; Lu, T.; Vogelbaum, M.A. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J. Neurooncol. 2007, 85, 39–47.
- Smith, D.; Shimamura, T.; Barbera, S.; Bejcek, B.E. NF-kappaB controls growth of glioblastomas/astrocytomas. Mol. Cell. Biochem. 2008, 307, 141–147.
- Cahill, K.E.; Morshed, R.A.; Yamini, B. Nuclear factor-kappaB in glioblastoma: Insights into regulators and targeted therapy. Neuro-Oncology 2016, 18, 329–339.
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro-Oncology 2015, 17, 45–52.
- Martinez-Olivera, R.; Datsi, A.; Stallkamp, M.; Koller, M.; Kohtz, I.; Pintea, B.; Gousias, K. Silencing of the nucleocytoplasmic shuttling protein karyopherin a2 promotes cell-cycle arrest and apoptosis in glioblastoma multiforme. Oncotarget 2018, 9, 33471–33481.
- Gousias, K.; Becker, A.J.; Simon, M.; Niehusmann, P. Nuclear karyopherin a2: A novel biomarker for infiltrative astrocytomas. J. Neurooncol. 2012, 109, 545–553.
- Gousias, K.; Niehusmann, P.; Gielen, G.H.; Simon, M. Karyopherin a2 and chromosome region maintenance protein 1 expression in meningiomas: Novel biomarkers for recurrence and malignant progression. J. Neurooncol. 2014, 118, 289–296.
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316.
- Biau, J.; Chautard, E.; Berthault, N.; de Koning, L.; Court, F.; Pereira, B.; Verrelle, P.; Dutreix, M. Combining the DNA Repair Inhibitor Dbait With Radiotherapy for the Treatment of High Grade Glioma: Efficacy and Protein Biomarkers of Resistance in Preclinical Models. Front. Oncol. 2019, 9, 549.
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102.
- Warters, R.L.; Hofer, K.G.; Harris, C.R.; Smith, J.M. Radionuclide toxicity in cultured mammalian cells: Elucidation of the primary site of radiation damage. Curr. Top. Radiat. Res. Q. 1978, 12, 389–407.
- Franken, N.A.; ten Cate, R.; Krawczyk, P.M.; Stap, J.; Haveman, J.; Aten, J.; Barendsen, G.W. Comparison of RBE values of high-LET alpha-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiat. Oncol. 2011, 6, 64.
- Zhu, P.; Zhu, J.J. Tumor treating fields: A novel and effective therapy for glioblastoma: Mechanism, efficacy, safety and future perspectives. Chin. Clin. Oncol. 2017, 6, 41.
- Gera, N.; Yang, A.; Holtzman, T.S.; Lee, S.X.; Wong, E.T.; Swanson, K.D. Tumor treating fields perturb the localization of septins and cause aberrant mitotic exit. PLoS ONE 2015, 10, e0125269.
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro-Oncology 2021, 23, 920–931.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Weller, M.; Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029.
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688.
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354.
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795.
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753.
- Costa, B.; Bendinelli, S.; Gabelloni, P.; Da Pozzo, E.; Daniele, S.; Scatena, F.; Vanacore, R.; Campiglia, P.; Bertamino, A.; Gomez-Monterrey, I.; et al. Human glioblastoma multiforme: p53 reactivation by a novel MDM2 inhibitor. PLoS ONE 2013, 8, e72281.
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196.
- Tien, A.C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786.
- Nguyen, L.V.; Searle, K.; Jerzak, K.J. Central nervous system-specific efficacy of CDK4/6 inhibitors in randomized controlled trials for metastatic breast cancer. Oncotarget 2019, 10, 6317–6322.
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238.
- Tong, H.; Zhao, K.; Zhang, J.; Zhu, J.; Xiao, J. YB-1 modulates the drug resistance of glioma cells by activation of MDM2/p53 pathway. Drug Des. Devel. Ther. 2019, 13, 317–326.
- Wu, Q.; Cao, Z.; Xiao, W.; Zhu, L.; Xie, Q.; Li, L.; Zhang, B.; Zhao, W. Study on Therapeutic Action and Mechanism of TMZ Combined with RITA Against Glioblastoma. Cell. Physiol. Biochem. 2018, 51, 2536–2546.
- Rouland, L.; Duplan, E.; Ramos Dos Santos, L.; Bernardin, A.; Katula, K.S.; Manfioletti, G.; Idbaih, A.; Checler, F.; Alves da Costa, C. Therapeutic potential of parkin as a tumor suppressor via transcriptional control of cyclins in glioblastoma cell and animal models. Theranostics 2021, 11, 10047–10063.
- Clark, P.A.; Bhattacharya, S.; Elmayan, A.; Darjatmoko, S.R.; Thuro, B.A.; Yan, M.B.; van Ginkel, P.R.; Polans, A.S.; Kuo, J.S. Resveratrol targeting of AKT and p53 in glioblastoma and glioblastoma stem-like cells to suppress growth and infiltration. J. Neurosurg. 2017, 126, 1448–1460.
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925.
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat Rev 2019, 80, 101896.
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442.
- Fan, H.C.; Chi, C.S.; Chang, Y.K.; Tung, M.C.; Lin, S.Z.; Harn, H.J. The Molecular Mechanisms of Plant-Derived Compounds Targeting Brain Cancer. Int. J. Mol. Sci. 2018, 19, 395.
- Wong, S.C.; Kamarudin, M.N.A.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950.
- Su, C.C.; Wang, M.J.; Chiu, T.L. The anti-cancer efficacy of curcumin scrutinized through core signaling pathways in glioblastoma. Int. J. Mol. Med. 2010, 26, 217–224.
- Karmakar, S.; Banik, N.L.; Ray, S.K. Curcumin suppressed anti-apoptotic signals and activated cysteine proteases for apoptosis in human malignant glioblastoma U87MG cells. Neurochem. Res. 2007, 32, 2103–2113.
- Doan, P.; Musa, A.; Candeias, N.R.; Emmert-Streib, F.; Yli-Harja, O.; Kandhavelu, M. Alkylaminophenol Induces G1/S Phase Cell Cycle Arrest in Glioblastoma Cells Through p53 and Cyclin-Dependent Kinase Signaling Pathway. Front. Pharmacol. 2019, 10, 330.
- Yeh, L.T.; Hsu, L.S.; Chung, Y.H.; Chen, C.J. Tectorigenin Inhibits Glioblastoma Proliferation by G0/G1 Cell Cycle Arrest. Medicina 2020, 56, 681.
- Alexiou, G.A.; Lazari, D.; Markopoulos, G.; Vartholomatos, E.; Hodaj, E.; Galani, V.; Kyritsis, A.P. Moschamine inhibits proliferation of glioblastoma cells via cell cycle arrest and apoptosis. Tumour Biol. 2017, 39, 1010428317705744.
- Wadhwa, E.; Nicolaides, T. Bromodomain Inhibitor Review: Bromodomain and Extra-terminal Family Protein Inhibitors as a Potential New Therapy in Central Nervous System Tumors. Cureus 2016, 8, e620.
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777.
- Jermakowicz, A.M.; Rybin, M.J.; Suter, R.K.; Sarkaria, J.N.; Zeier, Z.; Feng, Y.; Ayad, N.G. The novel BET inhibitor UM-002 reduces glioblastoma cell proliferation and invasion. Sci. Rep. 2021, 11, 23370.
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909.
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895.
- Gareev, I.; Beylerli, O.; Liang, Y.; Xiang, H.; Liu, C.; Xu, X.; Yuan, C.; Ahmad, A.; Yang, G. The Role of MicroRNAs in Therapeutic Resistance of Malignant Primary Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 740303.
- Wang, R.; Gurguis, C.I.; Gu, W.; Ko, E.A.; Lim, I.; Bang, H.; Zhou, T.; Ko, J.H. Ion channel gene expression predicts survival in glioma patients. Sci. Rep. 2015, 5, 11593.
- Gallia, G.L.; Holdhoff, M.; Brem, H.; Joshi, A.D.; Hann, C.L.; Bai, R.Y.; Staedtke, V.; Blakeley, J.O.; Sengupta, S.; Jarrell, T.C.; et al. Mebendazole and temozolomide in patients with newly diagnosed high-grade gliomas: Results of a phase 1 clinical trial. Neurooncol. Adv. 2021, 3, vdaa154.
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-Oncology 2011, 13, 974–982.
- Hu, Y.; Zhou, W.; Xue, Z.; Liu, X.; Feng, Z.; Zhang, Y.; Zhang, X.; Liu, X.; Li, W.; Zhang, Q.; et al. Thiabendazole inhibits glioblastoma cell proliferation and invasion targeting MCM2. J. Pharmacol. Exp. Ther. 2022, 380, 63–75.
- Zhou, X.; Liu, J.; Zhang, J.; Wei, Y.; Li, H. Flubendazole inhibits glioma proliferation by G2/M cell cycle arrest and pro-apoptosis. Cell Death Discov. 2018, 4, 18.
- Ren, L.W.; Li, W.; Zheng, X.J.; Liu, J.Y.; Yang, Y.H.; Li, S.; Zhang, S.; Fu, W.Q.; Xiao, B.; Wang, J.H.; et al. Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle. Acta Pharmacol. Sin. 2022, 43, 194–208.
- Weissenrieder, J.S.; Reed, J.L.; Green, M.V.; Moldovan, G.L.; Koubek, E.J.; Neighbors, J.D.; Hohl, R.J. The Dopamine D2 Receptor Contributes to the Spheroid Formation Behavior of U87 Glioblastoma Cells. Pharmacology 2020, 105, 19–27.
- Lin, W.Z.; Liu, Y.C.; Lee, M.C.; Tang, C.T.; Wu, G.J.; Chang, Y.T.; Chu, C.M.; Shiau, C.Y. From GWAS to drug screening: Repurposing antipsychotics for glioblastoma. J. Transl. Med. 2022, 20, 70.
- Papadopoulos, F.; Isihou, R.; Alexiou, G.A.; Tsalios, T.; Vartholomatos, E.; Markopoulos, G.S.; Sioka, C.; Tsekeris, P.; Kyritsis, A.P.; Galani, V. Haloperidol Induced Cell Cycle Arrest and Apoptosis in Glioblastoma Cells. Biomedicines 2020, 8, 595.
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; Group, A.L.-G.S. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401.
- Schipmann, S.; Muther, M.; Stogbauer, L.; Zimmer, S.; Brokinkel, B.; Holling, M.; Grauer, O.; Suero Molina, E.; Warneke, N.; Stummer, W. Combination of ALA-induced fluorescence-guided resection and intraoperative open photodynamic therapy for recurrent glioblastoma: Case series on a promising dual strategy for local tumor control. J. Neurosurg. 2020, 1–11.
- Jalili-Nik, M.; Abbasinezhad-Moud, F.; Sahab-Negah, S.; Maghrouni, A.; Etezad Razavi, M.; Khaleghi Ghadiri, M.; Stummer, W.; Gorji, A. Antitumor Effects of 5-Aminolevulinic Acid on Human Malignant Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 5596.
- Griffin, M.; Khan, R.; Basu, S.; Smith, S. Ion Channels as Therapeutic Targets in High Grade Gliomas. Cancers 2020, 12, 3068.
- Hemmerlein, B.; Weseloh, R.M.; Mello de Queiroz, F.; Knotgen, H.; Sanchez, A.; Rubio, M.E.; Martin, S.; Schliephacke, T.; Jenke, M.; Heinz Joachim, R.; et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 2006, 5, 41.
- Pollak, J.; Rai, K.G.; Funk, C.C.; Arora, S.; Lee, E.; Zhu, J.; Price, N.D.; Paddison, P.J.; Ramirez, J.M.; Rostomily, R.C. Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLoS ONE 2017, 12, e0172884.
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. 2015, 36, 9209–9213.
- Staudacher, I.; Jehle, J.; Staudacher, K.; Pledl, H.W.; Lemke, D.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PLoS ONE 2014, 9, e88164.
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. J. Neurosci. Res. 2004, 78, 224–234.
- Drucker, E.; Holzer, K.; Pusch, S.; Winkler, J.; Calvisi, D.F.; Eiteneuer, E.; Herpel, E.; Goeppert, B.; Roessler, S.; Ori, A.; et al. Karyopherin alpha2-dependent import of E2F1 and TFDP1 maintains protumorigenic stathmin expression in liver cancer. Cell Commun. Signal. 2019, 17, 159.
- Green, A.L.; Ramkissoon, S.H.; McCauley, D.; Jones, K.; Perry, J.A.; Hsu, J.H.; Ramkissoon, L.A.; Maire, C.L.; Hubbell-Engler, B.; Knoff, D.S.; et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro-Oncology 2015, 17, 697–707.
- Lassman, A.B.; Wen, P.Y.; van den Bent, M.J.; Plotkin, S.R.; Walenkamp, A.M.E.; Green, A.L.; Li, K.; Walker, C.J.; Chang, H.; Tamir, S.; et al. A Phase 2 Study of the Efficacy and Safety of Oral Selinexor in Recurrent Glioblastoma. Clin. Cancer Res. 2021, 28, 452–460.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
28 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No