 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandro Carrer | + 2071 word(s) | 2071 | 2020-09-16 05:54:29 | | | |
| 2 | Nora Tang | -75 word(s) | 1996 | 2020-09-24 07:46:01 | | | | |
| 3 | Nora Tang | -75 word(s) | 1996 | 2020-09-24 07:46:51 | | |
Video Upload Options
The carcinogenesis of pancreatic ductal adenocarcinoma (PDA) progresses according to multi-step evolution, whereby the disease acquires increasingly aggressive pathological features. Lineage-tracing experiments demonstrated that pancreatic cancerous lesions originate from acinar cells, a highly specialized cell type in the pancreatic epithelium. Acinar cells are polarized, pyramidal-shaped cells containing numerous acidophilic granules near the apical side. Those granules contain inactive proteases, which are activated and released into the tubular network upon activation. Primary acinar cells can survive in vitro as organoid-like 3D spheroids, which can transdifferentiate into cells with a clear ductal morphology in response to different cell- and non-cell-autonomous stimuli.
This event, termed acinar-to-ductal metaplasia, recapitulates the histological and molecular features of disease initiation.
1. Introduction
Pancreatic adenocarcinoma (PDA) is the most common form of pancreatic cancer, a disease that poses significant therapeutic challenges and accounts for more than 400,000 deaths worldwide [1]. The extremely poor survival rate of 9% with a five-year survival rate makes PDA the most lethal among major forms of cancer [2]. PDA remains asymptomatic for years and manifests at very late stages, often making the tumor unresectable [2][3]. A significant improvement of patient prognosis comes with earlier diagnoses, which are mostly anecdotal due to the poor understanding of disease initiation and the lack of biomarkers for early detection [4].
PDA progresses through a multi-step mechanism, where tumor formation is preceded by the formation of non-invasive pre-neoplastic lesions [5][6]. Genetic, epigenetic, and microenvironmental alterations are necessary for the formation of pre-malignant foci as well as for their progression to frank carcinoma. Understanding the cascade of events that leads to oncogenic transformation could illuminate means to prevent the onset of this deadly disease, or to enhance early diagnosis [4].
Dysplastic lesions are characterized by a distinctive duct-like morphology, which has led to the assumption that PDA arises from the expansion of mutated ductal cells. However, this notion has been increasingly disputed by several pieces of evidence since the early 1990s. More recently, a combination of lineage tracing experiments, genomic analyses of human specimens and in vitro examinations led to the notion that most pancreatic tumors originate from post-mitotic exocrine acinar cells (discussed below and in [7]).
2. Anatomy and Physiology of the Pancreas
Located in the retroperitoneum, the pancreas extends from the C-shaped curve of the duodenum (head), passes behind the stomach (body), and finishes at the hilum of the spleen (tail). Anatomical placement and gross anatomy of the organ are illustrated in Figure 1A.
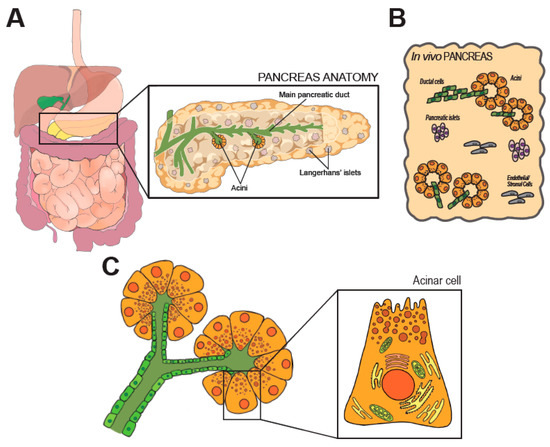
Figure 1. Anatomy and histology of the pancreas. In (A) anatomic organization of the GI tract. The pancreas is magnified on the right, along few representative structures within the organ. (B) shows the cellular heterogeneity of the pancreas, which is a composite of exocrine (acini), endocrine (Langherans’ islets) and stromal elements. (C) shows the modular units of the exocrine pancreas (acini). Morphology of an acinar cell is magnified on the right.
The developmentally mature pancreas is composed by a diverse arrangement of epithelial cells (acinar, ductal, the array of endocrine cells) and stromal cells [6], as schematically shown in Figure 1B. All epithelial cell types retain an elevated degree of plasticity in adult life and can transdifferentiate, a feature with significant pathophysiological and therapeutic ramifications [8].
The main pancreatic duct runs longitudinally through the extension of the organ, and branches in a tree of smaller conduits (Figure 1A) that are responsible to collect the digestive enzymes produced in the lobular units of the pancreas. Secretory units are organized in packed serous acinar glands, or acini, which form an anastomosis with the tubular network (Figure 1C). Each acinus is composed of post-mitotic epithelial cells, termed acinar cells (AC), and a few intercalated ductal cells that form the intra-acinar portion of the ductal system, along with one centroacinar cell that exhibits mixed traits [6]. ACs are polarized, pyramidal-shaped cells containing numerous acidophilic granules near the apical side. Those granules contain inactive proteases, which are activated and released into the tubular network upon activation. Exocytosis is promoted by elevation of circulating levels of cholecystokinin (CCK), gut-derived Secretin, or by direct vagal stimulation [9][10]. Early immunostaining examinations and recent analyses of individual cell transcriptomics (sc/snRNA-Seq) indicate the existence of distinct subpopulations of acinar cells that might reflect regional and/or functional differences [11][12]. The biological significance of the diversity in the exocrine compartment is difficult to justify [13]. In fact, the herd of ACs is histologically and morphologically indistinguishable. Some subsets may possess superior replicative capacity to support tissue homeostasis and repair [11]. However, ACs are long-living and the turnover of the pancreatic epithelium is limited. Thus, the presence of pluripotent-like cells in the adult pancreas is still debated [13].
3. Pancreatic Carcinogenesis
PDA tumors are frequently located in the head of the pancreas, and overhaul pancreatic tissue architecture, with extensive stromal deposition and over-representation of duct-like structures [6]. The reconstruction of tumor cells’ genomic history indicates that at least eight years span between the first oncogenic event and the detection of an overt PDA mass [14]. This clinically silent incubation period implies a typical late manifestation, which has led to a gap in the study of early stages of pancreatic carcinogenesis.
Genomic and histopathological analyses of either autoptic pancreata or early stage lesions or aggressive tumors only partially illuminate the transformative molecular events that lead to PDA [14][15][16][17]. In the early 2000s, the development of autochthonous mouse models of pancreatic tumorigenesis allowed the monitoring of disease progression since its inception [18][19], but only recent lineage-tracing experiments demonstrated that cancerous foci predominantly originate from metaplastic acinar cells, which undergo ductal transdifferentiation in response to environmental or oncogenic stress [20]. This tumor-initiating event is termed acinar-to-ductal metaplasia (ADM), and is normally a physiologic, reversible process that supports tissue repair. Temporary metaplasia occurs during pancreatic regeneration after injury. In mice, drug-induced inflammation causes extensive loss of acinar cells and drives temporary ADM in surviving cells to restore normal tissue histology after few days [21]. However, oncogenic inputs (both cell- and non-cell-autonomous) make ADM irreversible so that metaplastic cells engage a step-wise process that leads to the formation of low- and high-grade in situ dysplasia and eventually to metastatic carcinoma (Figure 2A) [5][22].
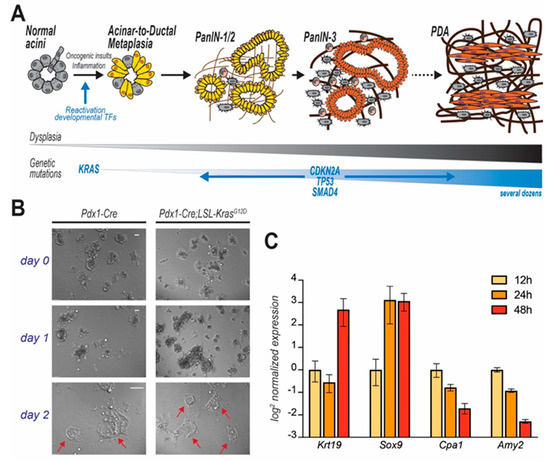
Figure 2. PDA progression and in vitro modelling of disease initiation. In (A) step-wise model of PDA progression. Upon mutation of KRAS, pancreatic acinar cells undergo acinar-to-ductal metaplasia (ADM), a cell transforming event which initiates the carcinogenic cascade. Cells progressively acquire dysplastic features and accrue genetic mutations. Acinar cell-derived pre-cancerous lesions are termed Pancreatic Intraepithelial Neoplasms (PanIN) that are defined by different histological grades and eventually evolve to carcinomas. In (B) acinar cells isolated from mice (genotypes indicated), immediately embedded in Matrigel and observed over time. Duct-like cystic structures (arrows) are observed after few days. In (C) time course analysis reveals that KC-derived primary acinar cells undergoing ADM ex vivo (Matrigel-embedded) upregulate duct-specific genes (Krt19 and Sox9), and suppress acinar-specific genes (Cpa1 and Amy2). Data for each gene are normalized to the first time point (12 h post isolation).
A variety of dysplastic precursors have been classified according to histopathological characteristics. Pancreatic intraepithelial neoplasia (PanIN) is the most commonly observed, and histological analysis of human specimens often reveals areas of acinar-to-ductal metaplasia in close proximity to low-grade PanIN foci [23][24], suggesting that the latter could in fact arise from the expansion of metaplastic cells.
Mutations in the coding sequence of the KRAS oncogene are nearly omnipresent in human PDA samples, but are also commonly observed in pre-neoplastic lesions [5]. Bioinformatic deconvolution of whole-genome analyses traced KRAS mutations very early in the oncogenic progression [14]. Strikingly, mice expressing the mutated form of KRAS (KrasG12D) in the pancreatic epithelium (Pdx1-Cre;LSL-KrasG12D-“KC mice”) spontaneously develop widespread ADM, which lead to the appearance of several PanIN foci by few weeks of age [18]. Current research shows that activating mutations of KRAS can exploit acinar cell plasticity to trigger ADM, which eventually progresses to form cancerous lesions upon additional genetic and epigenetic alterations [22][25][26]. Understanding the role of acinar cells in disease is of utmost importance due to their abundance in the parenchyma and their marked vulnerability to oncogenic insults.
4. Organotypic (3D) Acinar Cell Culture
Biochemical examination of acinar cells in vitro is essential to elucidate the cellular mechanisms of pancreatic carcinogenesis [27]. The protocol for the isolation and the culture of acinar cells has been optimized by several laboratories over the past decades [28][29][30][31], but still hinges on a light enzymatic digestion of minced pancreata to preserve the tri-dimensional organization of the acini. Digestion time is variable [29] and results in a heterogeneous mixture of microscopic tissue explants. Cocktails of Ser/Thr proteases inhibitors are added in order to prevent autodigestion of the explants (due to stress-induced release of proteases from acinar cells). A gradient sedimentation step enriches the preparation for acini, which sink after slow centrifugation, although that does not impede the accidental co-isolation of other cell populations. Finally, cell suspension is resuspended in Waymouth’s complete medium, or alternatively embedded in a collagen I matrix or Matrigel®. The short-term suspension culture of dispersed acini is preferred to study the biochemistry and pathophysiology of the exocrine pancreas [32]. A matrix scaffold can extend the in vitro culture period to up to 10 days and allows morphological and immunostaining analyses, but induces spontaneous ADM [28]. In particular, Matrigel® contains a population of growth factors that varies considerably across batches and can impact cell identity.
5. 3D Culture of ACs to Study Pancreatic Cancer Initiation
In light of the observation that PDA often originates from acinar cells that underwent ADM [20], the culture of dispersed acini has been adopted by cancer researchers to interrogate the molecular pathways leading to transdifferentiation and initiation of oncogenesis. Murine ex vivo cultures are excellent surrogates for in vivo models. When embedded in a 3D matrix (typically, collagen or Matrigel®), freshly isolated acinar cells organize in grape-like structures (acini), easily recognizable using an optic microscope. Both cell-intrinsic and -extrinsic factors can trigger ductal transdifferentiation (Figure 2B). Ex vivo, ADM is often initiated by addition of recombinant TGF or EGF, although that is not necessary when using growth factor-rich matrices (such as Matrigel®). During this process, acinar cells change morphology, but also lose their secretory function and markedly alter their gene expression profile, progressively increasing the expression of duct-specific genes, and reducing the levels of genes encoding for acinar proteases (Figure 2C). After a few days (variable), an elementary morphological examination reveals the presence of several duct-like, flat, circular structures, not originally present in the plate. Although a level of plasticity is innate in acinar cells, acini-to-duct conversion can be accelerated or suppressed by multiple factors that ultimately also either promote or inhibit pancreatic tumor initiation in vivo.
6. Final considerations
Acinar-to-ductal metaplasia is widespread in KC mice and gives origin to most precancerous lesions in model organisms. Isolation of primary acini and their transdifferentiation in vitro is a useful tool to decipher the network of events that lead to this permanent switch of cellular identity, which marks the initial step in pancreatic carcinogenesis.
It is increasingly clear that pancreatic acinar cells are a diverse bunch [11][12][13]. It is conceivable that one or more subsets of acinar cells might be endowed with superior tumor-initiation capacity. At the same time, regional differences in vivo (nutrient/oxygen supply, distance from hormone-producing cells) might impact acinar cell phenotype.
Tumorigenic driver mutations are prevalent in humans, yet only very few cells transform. In KC mice, oncogenic Kras expression is widespread in the pancreatic epithelium but only a few scattered pre-neoplastic lesions arise. What renders a subset of acinar cells susceptible to transformation while the remaining population appears refractory? This is a fundamental question in cancer biology. Notably, ex vivo cultures are valuable tools to address this question in PDA. Flow cytometry of cultured acinar cells may permit to discriminate and separate cells undergoing metaplasia in response to certain stimuli, from cells undergoing incomplete transdifferentiation or maintaining their acinar phenotype.
In this entry, the pivotal role of acinar cells in pancreatic cancer initiation has been outlined; however, several aspects of acinar cell biology remain elusive and a better understanding of the molecular steps leading to pancreatic cancer inception is critical to refine current prevention strategies and reduce the number of new cases.
References
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947.
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020.
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–23.
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040.
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081.
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249.
- Maitra, A.; Leach, S.D. Disputed Paternity: The Uncertain Ancestry of Pancreatic Ductal Neoplasia. Cancer Cell 2012, 22, 701–703.
- Cleveland, M.H.; Sawyer, J.M.; Afelik, S.; Jensen, J.; Leach, S.D. Exocrine ontogenies: On the development of pancreatic acinar, ductal and centroacinar cells. Semin. Cell Dev. Biol. 2012, 23, 711–719.
- Murtaugh, L.C.; Keefe, M.D. Regeneration and Repair of the Exocrine Pancreas. Annu. Rev. Physiol. 2015, 77, 229–249.
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. The role of Ca2+ in the pathophysiology of pancreatitis. J. Physiol. 2014, 592, 269–280.
- Wollny, D.; Zhao, S.; Everlien, I.; Lun, X.; Brunken, J.; Brüne, D.; Ziebell, F.; Tabansky, I.; Weichert, W.; Marciniak-Czochra, A.; et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev. Cell 2016, 39, 289–301.
- Tosti, L.; Hang, Y.; Trefzer, T.; Steiger, K.; Ten, F.W.; Lukassen, S.; Ballke, S.; Kuehl, A.A.; Spieckermann, S.; Bottino, R.; et al. Single nucleus RNA sequencing maps acinar cell states in a human pancreas cell atlas. bioRxiv 2019, 733964.
- Jiang, Z.; White, R.A.; Wang, T.C. Adult Pancreatic Acinar Progenitor-like Populations in Regeneration and Cancer. Trends Mol. Med. 2020, 26, 758–767.
- Makohon-Moore, A.P.; Matsukuma, K.; Zhang, M.; Reiter, J.G.; Gerold, J.M.; Jiao, Y.; Sikkema, L.; Attiyeh, M.A.; Yachida, S.; Sandone, C.; et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018, 561, 201–205.
- Iacobuzio-Donahue, C.A.; Michael, C.; Baez, P.; Kappagantula, R.; Hooper, J.E.; Hollman, T.J. Cancer biology as revealed by the research autopsy. Nat. Rev. Cancer 2019, 19, 686–697.
- Kong, B.; Bruns, P.; Behler, N.A.; Chang, L.; Schlitter, A.M.; Cao, J.; Gewies, A.; Ruland, J.; Fritzsche, S.; Valkovskaya, N.; et al. Dynamic landscape of pancreatic carcinogenesis reveals early molecular networks of malignancy. Gut 2018, 67, 146–156.
- Matsuda, Y.; Furukawa, T.; Yachida, S.; Nishimura, M.; Seki, A.; Nonaka, K.; Aida, J.; Takubo, K.; Ishiwata, T.; Kimura, W.; et al. The prevalence and clinicopathological characteristics of high-grade pancreatic intraepithelial neoplasia autopsy study evaluating the entire pancreatic parenchyma. Pancreas 2017, 46, 658–664.
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450.
- DeCant, B.T.; Principe, D.R.; Guerra, C.; Pasca di Magliano, M.; Grippo, P.J. Utilizing past and present mouse systems to engineer more relevant pancreatic cancer models. Front. Physiol. 2014, 5, 464.
- Kopp, J.L.; Von Figura, G.; Mayes, E.; Liu, F.; Dubois, C.L.; Iv, J.P.M.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750.
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739.
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304.
- McCarthy, D.M.; Brat, D.J.; Wilentz, R.E.; Yeo, C.J.; Cameron, J.L.; Kern, S.E.; Hruban, R.H. Pancreatic intraepithelial neoplasia and infiltrating adenocarcinoma: Analysis of progression and recurrence by DPC4 immunohistochemical labeling. Hum. Pathol. 2001, 32, 638–642.
- Shi, C.; Klein, A.P.; Goggins, M.; Maitra, A.; Canto, M.; Ali, S.; Schulick, R.; Palmisano, E.; Hruban, R.H. Increased Prevalence of Precursor Lesions in Familial Pancreatic Cancer Patients. Clin. Cancer Res. 2009, 15, 7737–7743.
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2005, 1756, 97–101.
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229.
- Sphyris, N.; Logsdon, C.D.; Harrison, D.J. Improved retention of zymogen granules in cultured murine pancreatic acinar cells and induction of acinar-ductal transdifferentiation in vitro. Pancreas 2005, 30, 148–157.
- Gout, J.; Pommier, R.M.; Vincent, D.F.; Kaniewski, B.; Martel, S.; Valcourt, U.; Bartholin, L. Isolation and culture of mouse primary pancreatic acinar cells. J. Vis. Exp. 2013, 1–8.
- Fleming Martinez, A.K.; Storz, P. Mimicking and Manipulating Pancreatic Acinar-to-Ductal Metaplasia in 3-dimensional Cell Culture. J. Vis. Exp. 2019.
- Pinho, A.V.; Rooman, I.; Reichert, M.; De Medts, N.; Bouwens, L.; Rustgi, A.K.; Real, F.X. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut 2011, 60, 958–966.
- Houbracken, I.; de Waele, E.; Lardon, J.; Ling, Z.; Heimberg, H.; Rooman, I.; Bouwens, L. Lineage tracing evidence for transdifferentiation of acinar to duct cells and plasticity of human pancreas. Gastroenterology 2011, 141, 731–741.e4.
- Bläuer, M.; Nordback, I.; Sand, J.; Laukkarinen, J. A novel explant outgrowth culture model for mouse pancreatic acinar cells with long-term maintenance of secretory phenotype. Eur. J. Cell Biol. 2011, 90, 1052–1060.

