 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hidenori Suzuki | + 3577 word(s) | 3577 | 2022-03-14 03:27:43 | | | |
| 2 | Bruce Ren | Meta information modification | 3577 | 2022-03-28 03:51:09 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3577 | 2022-03-28 04:52:37 | | |
Video Upload Options
Delayed cerebral ischemia (DCI) remains a challenging but very important condition, because DCI is preventable and treatable for improving functional outcomes after aneurysmal subarachnoid hemorrhage (SAH). The pathologies underlying DCI are multifactorial. Classical approaches to DCI focus exclusively on preventing and treating the reduction of blood flow supply. However, recently, glutamate-mediated neuroelectric disruptions, such as excitotoxicity, cortical spreading depolarization and seizures, and epileptiform discharges, have been reported to occur in high frequencies in association with DCI development after SAH. Each of the neuroelectric disruptions can trigger the other, which augments metabolic demand. If increased metabolic demand exceeds the impaired blood supply, the mismatch leads to relative ischemia, resulting in DCI. The neuroelectric disruption also induces inverted vasoconstrictive neurovascular coupling in compromised brain tissues after SAH, causing DCI. Although glutamates and the receptors may play central roles in the development of excitotoxicity, cortical spreading ischemia and epileptic activity-related events, more studies are needed to clarify the pathophysiology and to develop novel therapeutic strategies for preventing or treating neuroelectric disruption-related DCI after SAH.
1. Introduction
2. Glutamate
2.1. Signaling via Glutamates
2.2. Major Glutamate Receptors
2.3. Glutamate in Blood Vessels
2.4. Glutamate in SAH
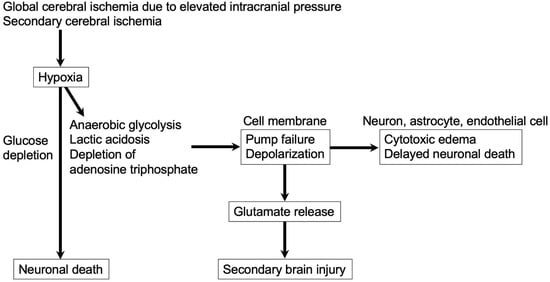
2.5. Glutamate and Inverse Neurovascular Coupling after SAH
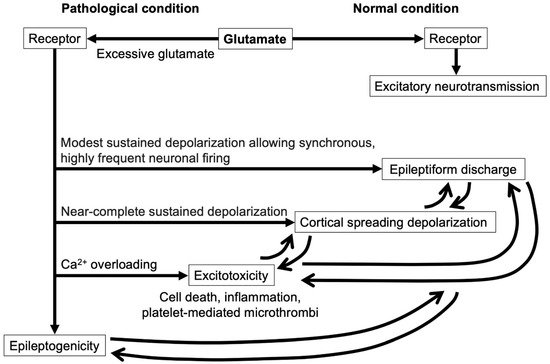
3. Excitotoxity in Post-SAH Ischemic Brain
3.1. Glutamate Receptors and Ions in Excitotoxicity
3.2. Relationships among Inflammation, Microthrombus, and Excitotoxity
References
- Kanamaru, H.; Kawakita, F.; Asada, R.; Miura, Y.; Shiba, M.; Toma, N.; Suzuki, H.; pSEED group. Prognostic factors varying with age in patients with aneurysmal subarachnoid hemorrhage. J. Clin. Neurosci. 2020, 76, 118–125.
- Suzuki, H.; Fujimoto, M.; Kawakita, F.; Liu, L.; Nakatsuka, Y.; Nakano, F.; Nishikawa, H.; Okada, T.; Kanamaru, H.; Imanaka-Yoshida, K.; et al. Tenascin-C in brain injuries and edema after subarachnoid hemorrhage: Findings from basic and clinical studies. J. Neurosci. Res. 2020, 98, 42–56.
- Suzuki, H. Inflammation: A good research target to improve outcomes of poor-grade subarachnoid hemorrhage. Transl. Stroke Res. 2019, 10, 597–600.
- Geraghty, J.R.; Testai, F.D. Delayed Cerebral Ischemia after Subarachnoid Hemorrhage: Beyond Vasospasm and Towards a Multifactorial Pathophysiology. Curr. Atheroscler. Rep. 2017, 19, 50.
- Suzuki, H.; Kanamaru, H.; Kawakita, F.; Asada, R.; Fujimoto, M.; Shiba, M. Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol. Histopathol. 2021, 36, 143–158.
- Suzuki, H. What is early brain injury? Transl. Stroke Res. 2015, 6, 1–3.
- Xiao, M.; Li, Q.; Feng, H.; Zhang, L.; Chen, Y. Neural vascular mechanism for the cerebral blood flow auto-regulation after hemorrhagic stroke. Neural Plast. 2017, 2017, 5819514.
- Dreier, J.P.; Major, S.; Pannek, H.W.; Woitzik, J.; Scheel, M.; Wiesenthal, D.; Martus, P.; Winkler, M.K.; Hartings, J.A.; Fabricius, M.; et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain 2012, 135, 259–275.
- Nakano, F.; Liu, L.; Kawakita, F.; Kanamaru, H.; Nakatsuka, Y.; Nishikawa, H.; Okada, T.; Shiba, M.; Suzuki, H. Morphological characteristics of neuronal death after experimental subarachnoid hemorrhage in mice using double immunoenzymatic technique. J. Histochem. Cytochem. 2019, 67, 919–930.
- Chung, D.Y.; Oka, F.; Ayata, C. Spreading depolarizations: A therapeutic target against delayed cerebral ischemia after subarachnoid hemorrhage. J. Clin. Neurophysiol. 2016, 33, 196–202.
- Zafar, S.F.; Postma, E.N.; Biswal, S.; Boyle, E.J.; Bechek, S.; O’Connor, K.; Shenoy, A.; Kim, J.; Shafi, M.S.; Patel, A.B.; et al. Effect of epileptiform abnormality burden on neurologic outcome and antiepi-leptic drug management after subarachnoid hemorrhage. Clin. Neurophysiol. 2018, 129, 2219–2227.
- Helbok, R.; Kofler, M.; Schiefecker, A.J.; Gaasch, M.; Rass, V.; Pfausler, B.; Beer, R.; Schmutzhard, E. Clinical Use of Cerebral Microdialysis in Patients with Aneurysmal Subarachnoid Hemorrhage—State of the Art. Front. Neurol. 2017, 8, 565.
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953.
- Castillo, J.; Loza, M.I.; Mirelman, D.; Brea, J.; Blanco, M.; Sobrino, T.; Campos, F. A novel mechanism of neuroprotection: Blood glutamate grabber. J. Cereb. Blood Flow Metab. 2016, 36, 292–301.
- Baranovic, J. AMPA receptors in the synapse: Very little space and even less time. Neuropharmacology 2021, 196, 108711.
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322.
- Casillas-Espinosa, P.M.; Powell, K.L.; O’Brien, T.J. Regulators of synaptic transmission: Roles in the pathogenesis and treatment of epilepsy. Epilepsia 2020, 53 (Suppl. S9), 41–58.
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188.
- Serwach, K.; Gruszczynska-Biegala, J. STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2289.
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542.
- Scheefhals, N.; MacGillavry, H.D. Functional organization of postsynaptic glutamate receptors. Mol. Cell. Neurosci. 2018, 91, 82–94.
- Casillas-Espinosa, P.M.; Ali, I.; O’Brien, T.J. Neurodegenerative pathways as targets for acquired epilepsy therapy development. Epilepsia Open 2020, 5, 138–154.
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108.
- Isaac, J.T.; Ashby, M.C.; McBain, C.J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 2007, 54, 859–871.
- Diering, G.H.; Huganir, R.L. The AMPA receptor code of synaptic plasticity. Neuron 2018, 100, 314–329.
- Chang, D.T.W.; Reynolds, I.J. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience 2006, 141, 727–736.
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318.
- Chen, T.; Zhu, J.; Wang, Y.H. RNF216 mediates neuronal injury following experimental subarachnoid hemorrhage through the Arc/Arg3.1-AMPAR pathway. FASEB J. 2020, 34, 15080–15092.
- Chong, Z.Z.; Li, F.; Maiese, K. Group I metabotropic receptor neuroprotection requires Akt and its substrates that govern FOXO3a, Bim, and beta-catenin during oxidative stress. Curr. Neurovasc. Res. 2006, 3, 107–117.
- Xu, W.; Wong, T.P.; Chery, N.; Gaertner, T.; Wang, Y.T.; Baudry, M. Calpain-mediated mGluR1alpha truncation: A key step in excitotoxicity. Neuron 2007, 53, 399–412.
- Marrannes, R.; Willems, R.; De Prins, E.; Wauquier, A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988, 457, 226–240.
- Pin, J.P.; Duvoisin, R. The metabotropic glutamate receptors: Structure and functions. Neuropharmacology 1995, 34, 1–26.
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402.
- Sun, Y.; Feng, X.; Ding, Y.; Li, M.; Yao, J.; Wang, L.; Gao, Z. Phased Treatment Strategies for Cerebral Ischemia Based on Glutamate Receptors. Front. Cell. Neurosci. 2019, 13, 168.
- Bruno, V.; Battaglia, G.; Copani, A.; Giffard, R.G.; Raciti, G.; Raffaele, R.; Shinozaki, H.; Nicoletti, F. Activation of class II or III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur. J. Neurosci. 1995, 7, 1906–1913.
- Corti, C.; Battaglia, G.; Molinaro, G.; Riozzi, B.; Pittaluga, A.; Corsi, M.; Mugnaini, M.; Nicoletti, F.; Bruno, V. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J. Neurosci. 2007, 27, 8297–8308.
- Matsui, K.; Jahr, C.E. Differential control of synaptic and ectopic vesicular release of glutamate. J. Neurosci. 2004, 24, 8932–8939.
- Parfenova, H.; Fedinec, A.; Leffler, C.W. Ionotropic glutamate receptors in cerebral microvascular endothe-lium are functionally linked to heme oxygenase. J. Cereb. Blood Flow Metab. 2003, 23, 190–197.
- Brand-Schieber, E.; Lowery, S.L.; Werner, P. Select ionotropic glutamate AMPA/kainate receptors are expressed at the astrocyte-vessel interface. Brain Res. 2004, 1007, 178–182.
- Fergus, A.; Lee, K.S. Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res. 1997, 754, 35–45.
- Tso, M.K.; Macdonald, R.L. Subarachnoid hemorrhage: A review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 2014, 5, 174–189.
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-mediated blood-brain barrier opening: Implications for neuroprotection and drug delivery. J. Neurosci. 2016, 36, 7727–7739.
- Collard, C.D.; Park, K.A.; Montalto, M.C.; Alapati, S.; Buras, J.A.; Stahl, G.L.; Colgan, S.P. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J. Biol. Chem. 2002, 277, 14801–14811.
- András, I.E.; Deli, M.A.; Veszelka, S.; Hayashi, K.; Hennig, B.; Toborek, M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J. Cereb. Blood Flow Metab. 2007, 27, 1431–1443.
- Pula, G.; Krause, M. Role of Ena/VASP proteins in homeostasis and disease. Handb. Exp. Pharmacol. 2008, 186, 39–65.
- Mulligan, S.J.; MacVicar, B.A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199.
- Rancillac, A.; Rossier, J.; Guille, M.; Tong, X.K.; Geoffroy, H.; Amatore, C.; Arbault, S.; Hamel, E.; Cauli, B. Glutamatergic control of microvascular tone by distinct GABA neurons in the cerebellum. J. Neurosci. 2006, 26, 6997–7006.
- Kanamaru, H.; Suzuki, H. Potential therapeutic molecular targets for blood-brain barrier disruption after subarachnoid hemorrhage. Neural Regen. Res. 2019, 14, 1138–1143.
- Van Lieshout, J.H.; Dibué-Adjei, M.; Cornelius, J.F.; Slotty, P.J.; Schneider, T.; Restin, T.; Boogaarts, H.D.; Steiger, H.J.; Petridis, A.K.; Kamp, M.A. An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 2018, 41, 917–930.
- Mindt, S.; Tokhi, U.; Hedtke, M.; Groß, H.J.; Hänggi, D. Mass spectrometry-based method for quantification of nimodipine and glutamate in cerebrospinal fluid. Pilot study with patients after aneurysmal subarachnoid haemorrhage. J. Clin. Pharm. Ther. 2020, 45, 81–87.
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160.
- Bell, J.D.; Thomas, T.C.; Lass, E.; Ai, J.; Wan, H.; Lifshitz, J.; Baker, A.J.; Macdonald, R.L. Platelet-mediated changes to neuronal glutamate receptor expression at sites of microthrombosis following experimental subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1424–1431.
- Feng, D.; Wang, W.; Dong, Y.; Wu, L.; Huang, J.; Ma, Y.; Zhang, Z.; Wu, S.; Gao, G.; Qin, H. Ceftriaxone allevi-ates early brain injury after subarachnoid hemorrhage by increasing excitatory amino acid transporter 2 expression via the PI3K/Akt/NF-κB signaling pathway. Neuroscience 2014, 268, 21–32.
- Jung, C.S.; Lange, B.; Zimmermann, M.; Seifert, V. CSF and Serum Biomarkers Focusing on Cerebral Vasospasm and Ischemia after Subarachnoid Hemorrhage. Stroke Res. Treat. 2013, 2013, 560305.
- Wang, H.B.; Wu, Q.J.; Zhao, S.J.; Hou, Y.J.; Li, H.X.; Yang, M.F.; Wang, B.J.; Sun, B.L.; Zhang, Z.Y. Early high cerebrospinal fluid glutamate: A potential predictor for delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. ACS Omega 2020, 5, 15385–15389.
- Wu, C.T.; Wen, L.L.; Wong, C.S.; Tsai, S.Y.; Chan, S.M.; Yeh, C.C.; Borel, C.O.; Cherng, C.H. Temporal changes in glutamate, glutamate transporters, basilar arteries wall thickness, and neuronal variability in an experimental rat model of subarachnoid hemorrhage. Anesth. Analg. 2011, 112, 666–673.
- Zhang, Z.; Liu, J.; Fan, C.; Mao, L.; Xie, R.; Wang, S.; Yang, M.; Yuan, H.; Yang, X.; Sun, J.; et al. The GluN1/GluN2B NMDA receptor and metabotropic glutamate receptor 1 negative allosteric modulator has enhanced neuroprotection in a rat subarachnoid hemorrhage model. Exp. Neurol. 2018, 301, 13–25.
- Ahmadabad, R.A.; Ghadiri, M.K.; Gorji, A. The role of Toll-like receptor signaling pathways in cerebrovascular disorders: The impact of spreading depolarization. J. Neuroinflamm. 2020, 17, 108.
- Kramer, D.R.; Fujii, T.; Ohiorhenuan, I.; Liu, C.Y. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J. Clin. Neurosci. 2016, 24, 22–27.
- Balbi, M.; Koide, M.; Wellman, G.C.; Plesnila, N. Inversion of neurovascular coupling after subarachnoid hemorrhage in vivo. J. Cereb. Blood Flow Metab. 2017, 37, 3625–3634.
- Amantea, D.; Bagetta, G. Excitatory and inhibitory amino acid neurotransmitters in stroke: From neurotoxicity to ischemic tolerance. Curr. Opin. Pharmacol. 2017, 35, 111–119.
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668.
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770.
- Nicolo, J.-P.; Chen, Z.; Moffat, B.; Wright, D.K.; Sinclair, B.; Glarin, R.; Neal, A.; Thijs, V.; Seneviratne, U.; Yan, B.; et al. Study protocol for a phase II randomised, double-blind, placebo-controlled trial of perampanel as an antiepileptogenic treatment following acute stroke. BMJ Open 2021, 11, e043488.
- Pulsinelli, W.; Sarokin, A.; Buchan, A. Antagonism of the NMDA and non-NMDA receptors in global versus focal brain ischemia. Prog. Brain Res. 1993, 96, 125–135.
- Pellegrini-Giampietro, D.E.; Zukin, R.S.; Bennett, M.V.; Cho, S.; Pulsinelli, W.A. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc. Natl. Acad. Sci. USA 1992, 89, 10499–10503.
- Giffard, R.G.; Monyer, H.; Christine, C.W.; Choi, D.W. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990, 506, 339–342.
- Kim, Y.H.; Kim, E.Y.; Gwag, B.J.; Sohn, S.; Koh, J.Y. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: Mediation by free radicals. Neuroscience 1999, 89, 175–182.
- Zhang, Y.; Wang, H.; Li, J.; Dong, L.; Xu, P.; Chen, W.; Neve, R.L.; Volpe, J.J.; Rosenberg, P.A. Intracellular zinc release and ERK phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J. Biol. Chem. 2006, 281, 9460–9470.
- Yeh, S.C.; Tsai, F.Y.; Chao, H.R.; Tsou, T.C. Zinc ions induce inflammatory responses in vascular endothe-lial cells. Bull. Environ. Contam. Toxicol. 2011, 87, 113–116.
- Sumagin, R.; Lomakina, E.; Sarelius, I.H. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H969–H977.
- Skowrońska, K.; Obara-Michlewska, M.; Zielińska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309.
- Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20, 2450.
- Bradley, S.J.; Challiss, R.A.J. G protein-coupled receptor signalling in astrocytes in health and disease: A focus on metabotropic glutamate receptors. Biochem. Pharmacol. 2012, 84, 249–259.
- Anrather, J.; Iadecola, C. Inflammation and stroke: An overview. Neurotherapeutics 2016, 13, 661–670.
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142.
- Wang, J.; Liang, J.; Deng, J.; Liang, X.; Wang, K.; Wang, H.; Qian, D.; Long, H.; Yang, K.; Qi, S. Emerging role of microglia-mediated neuroinflammation in epilepsy after subarachnoid hemorrhage. Mol. Neurobiol. 2021, 58, 2780–2791.
- Friedrich, V.; Flores, R.; Muller, A.; Sehba, F.A. Luminal platelet aggregates in functional deficits in parenchymal vessels after subarachnoid hemorrhage. Brain Res. 2010, 1354, 179–187.
- McBride, D.W.; Blackburn, S.L.; Peeyush, K.T.; Matsumura, K.; Zhang, J.H. The Role of Thromboinflammation in Delayed Cerebral Ischemia after Subarachnoid Hemorrhage. Front. Neurol. 2017, 8, 555.
- Gautam, D.; Tiwari, A.; Chaurasia, R.N.; Dash, D. Glutamate induces synthesis of thrombogenic peptides and extracellular vesicle release from human platelets. Sci. Rep. 2019, 9, 8346.

