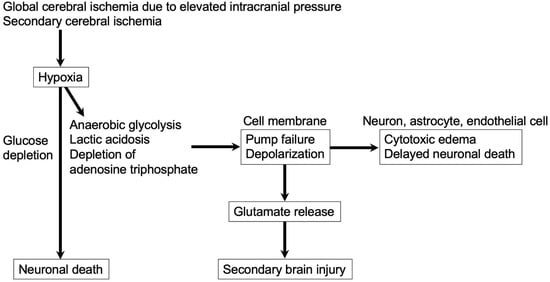
Excitotoxity contributes to both EBI and DCI after aneurysmal SAH. In EBI, massive aneurysmal rupture causes severe elevation of ICP, followed by transient cerebral circulation arrest, which leads to cessation of neuronal electrical activity within seconds, mitochondrial dysfunction associated with decreased production of adenosine triphosphate to deteriorate the energy state and to disrupt the Na
+-K
+ pump, and ion homeostasis, resulting in disturbed membrane ion gradients (depolarization), Ca
2+ influx, and extracellular release of a large amount of glutamates from depolarized nerve terminals and astrocytes within minutes [
61]. In DCI, secondary cerebral ischemia also triggers excessive glutamate releases. Massive releases of glutamates over-activate AMPA, NMDA, and kainate receptors on neurons, as well as other cellular components of the neurovascular unit, causing excessive intracellular Ca
2+ entry as the primary mediator of excitotoxicity through the receptors, which is augmented by Ca
2+ releases from endoplasmic reticulum via activation of mGluRs [
13,
61]. Intracellular Ca
2+ overload induces further release of glutamates and overactivation of multiple Ca
2+-dependent enzymes such as calpains, other proteases, protein kinases, calcineurins, endonucleases, phospholipases A
2, and xanthine oxidases [
13]. The activation of theses enzymes impairs mitochondrial energy production and causes increased production of ROS to promote lipid peroxidation, membrane failure, and cell damage, as well as alterations in the organization of the cytoskeleton, activation of genetic signals leading to cell death, and an increase in expressions of immediate early genes [
13,
33,
62]. Cellular damage also causes further glutamate releases [
13]. In addition, Ca
2+ influx activates neuronal NO synthases to produce NO, which may be involved in both normal neuronal signaling and free-radical-mediated glutamate excitotoxicity harnessed by macrophages [
13,
63]. Ca
2+ overload and oxidative stress, including NO, cause apoptosis and necrosis, which can occur caspase-dependently or -independently [
13]. Glutamates also increase deoxyribonucleic acid binding of the redox-regulated transcription factors, nuclear factor-κB, and activating protein 1, as well as upregulate the immediate early gene, c-fos, leading to glutamate-induced apoptosis or necrosis [
33]. Glutamate levels in cerebrospinal fluid and peripheral blood have been reported to be positively correlated with infarct size, infarct growth, and functional outcomes in clinical settings [
64]. In contrast, the neuroprotective neurotransmitter GABA rapidly and transiently increases in the extracellular space like most neurotransmitters, but the expressions of both GABA-A and GABA-B receptors decrease after cerebral ischemia to cause impaired GABA-mediated neurotransmission, which contributes to ongoing neuronal excitability and possibly to neuronal death [
61]. Excitotoxic neuronal death is not a uniform event but, rather, a continuum of necrotic, apoptotic, and autophagic morphologies [
33].
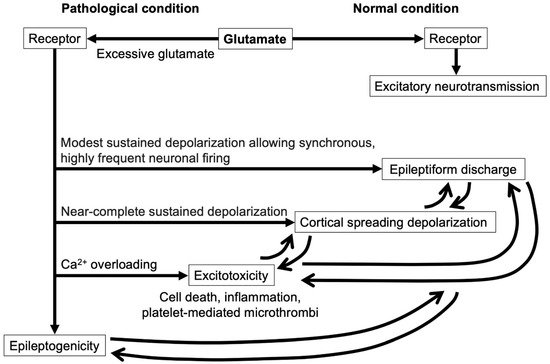
Excitotoxicity is composed of two components: the first one is an acute, intracellular influx of Na
+ and chloride ions followed by water influx, resulting in cell swelling, tissue edema and, consequently, impaired perfusion of the surrounding brain tissues, even in the absence of extracellular Ca
2+; the second one is Ca
2+-dependent delayed cellular degeneration [
62]. In contrast to neuronal swelling, which developed immediately after excessive glutamate exposure, delayed neuronal death was observed 24 h post-glutamate exposure and was abolished by the removal of Ca
2+ while potentiated by the addition of Ca
2+ [
18]. The removal of Na
+ from the culture medium prior to glutamate exposure prevented neuronal swelling but had no effects on delayed neuronal death [
18].
3.1. Glutamate Receptors and Ions in Excitotoxicity
Although the NMDA receptor was initially considered to be a critical mediator in focal cerebral ischemia, subsequent studies support a more central role for AMPA receptors in hippocampal injuries associated with global cerebral ischemia [
65]. AMPA receptors mediate Na
+ influx and therefore can contribute to excitotoxic Ca
2+ overload and neuronal death [
13]. Although AMPA receptors lack direct linkage to NO synthases and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, AMPA receptors participate substantially in brain damage after focal and global ischemia; in global ischemia, AMPA receptors typically contribute more than NMDA receptors to delayed death of selectively vulnerable neurons, possibly due to upregulation of Ca
2+-permeable AMPA receptors [
13]. Ischemia upregulates Ca
2+-permeable AMPA receptors in vulnerable neuronal populations, which constitute a dominant route for toxic Ca
2+/zinc ion (Zn
2+) entry [
66].
In addition to Na
+ and Ca
2+, H
+ participates in excitotoxicity. In ischemic brain tissues, extracellular pH typically drops within minutes toward 6.5 or lower due to anaerobic glycolysis for resynthesis of adenosine triphosphate, and an increase in extracellular H
+ attenuates NMDA receptor channel openings and NADPH oxidase 2 activity, resulting in reduced NMDA receptor-mediated excitotoxicity [
67]. However, ischemic acidosis is itself cytotoxic to both neurons and glia by enhancing neurotoxic Ca
2+ overload via the gating of acid-sensing ion channels and by being accompanied by an increase in intracellular Zn
2+ [
13]. Relatively low concentrations (≈20 μM) of Zn
2+ elicit apoptotic neuronal death, while higher concentrations (50–100 μM) of Zn
2+ cause neuronal death, with the characteristics of necrosis [
68]. In addition to promoting neuronal death, intracellular release of excitotoxic Zn
2+ contributes to the death of adjacent non-neuronal cells such as astrocytes, oligodendroglia, and capillary endothelial cells in ischemic brain tissues [
13]. An intracellular increase in Zn
2+ is considered to mediate peroxynitrite-induced death through activation of extracellular signal-regulated kinases 1/2 and arachidonate 12-lipoxygenases and thereby further ROS generation [
69], and to upregulate intercellular adhesion molecule-1 expression in vascular endothelial cells, promoting leukocyte attraction and microvascular leakage [
70,
71]. Several other membrane channels may be activated in part as a result of overstimulation of glutamate receptors and can contribute to toxic Ca
2+/Zn
2+ overload and other ionic derangements in ischemic brain tissues [
13].
Although excitotoxicity was originally described as specific to neurons, oligodendrocytes and astrocytes also suffer excitotoxic injury and death [
13]. Astrocytes are less insensitive to excitotoxicity compared with oligodendrocytes: this is because most astrocytes express AMPA receptors and mGluRs, but NMDA receptors and Ca
2+-permeable AMPA receptors are generally not abundant [
72,
73,
74]. However, as astrocytes are vulnerable to Zn
2+ or H
+-induced damages, astrocytic death may increase secondary to excitotoxicity occurring in nearby neurons or oligodendrocytes [
13].
3.2. Relationships among Inflammation, Microthrombus, and Excitotoxity
Excitotoxicity per se triggers and augments inflammatory reactions to continue destroying brain tissues. The neurovascular unit-constituent cells release cytokines and chemokines, recruiting leukocytes to the evolving ischemic region over hours to days, and advance microvascular damage and oxidative stress [
75,
76]. Inducible NO synthases are induced in infiltrating neutrophils and endothelial cells in ischemic brain tissues to produce NO and to synergize oxidatively with superoxide emanating from neutrophil NADPH oxidase 2 and endothelial NADPH oxidase 4, aggravating brain damage [
13]. Activated microglia are a significant source of redundant extracellular glutamates that induce excitotoxic neuronal death [
33]. Although microglia-mediated neuroinflammation in EBI may have impacts on neuronal excitotoxicity, BBB disruption, and the further changes of immune responses, all of which may lower the seizure threshold, activated microglia themselves may promote epilepsy development independent of the inflammatory responses [
77].
In DCI after SAH, platelet aggregates are also observed to be associated with or without focal microvascular constriction and to be extravasated into the brain parenchyma by platelet-mediated release of collagenase and subsequent depletion of collagen IV in vessel walls [
78]. The extravasated platelet aggregates or platelet-mediated microthrombi are reported not only to propagate pro-inflammatory signaling [
79] but also to release glutamates, causing excitotoxic brain injuries [
52]. Although glutamate does not cross the BBB, BBB disruption at sites of microthrombi or extravasated platelets that release glutamates during their lysis or aggregation may allow neurons to be exposed to excessive glutamates [
52]. Platelets have dense granules, carrying a considerable amount of glutamates, and also express glutamate receptors on their surface [
80]. Excessive glutamate is reported to induce platelet activation and synthesis of thrombogenic peptides, plasminogen activator inhibitor-1, and hypoxia-inducible factor-2α from pre-existing messenger ribonucleic acids in anucleate platelets, which are mediated mostly through AMPA receptors [
80].