 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | H.P. Vasantha Rupasinghe | + 1522 word(s) | 1522 | 2020-09-21 10:22:11 | | | |
| 2 | Camila Xu | -67 word(s) | 1455 | 2020-09-23 10:34:32 | | | | |
| 3 | Camila Xu | Meta information modification | 1455 | 2020-10-26 10:06:21 | | |
Video Upload Options
Gastrointestinal (GI) cancer is a heterogeneous cancer that tends to occur in the more common sporadic forms rather than the rare inherited forms. The process of initiation and formation of neoplastic cells in the GI tract can be classified into four main mechanisms: (i) inherited transmission of mutations; (ii) exposure to different carcinogens; (iii) chronic inflammatory conditions/microbial dysbiosis; and (iv) sporadic mutations and epigenetic changes.
1. Hereditary GI Cancers
Hereditary GI cancers represent a phenotypically diverse group of diseases involving malignant tumors of the digestive tract, extra-GI cancers, and benign abnormalities characterized by inherited genetic mutations transmitted from parent to child. However, no more than 3%–5% of GI cancers have shown a clear hereditary basis [1]. The esophagus, stomach, colon, small intestine, and pancreas have been identified as the organs most likely to inherit germline mutations [2]. The best known inherited malignant tumors are associated with the GI tract, representing monogenic hereditary diseases that result from mutations in a single gene [3]. Despite the specific differences in the genes involved, inherited GI cancers share a common set of characteristics: (i) the majority of GI cancers are detectable in the early stages of life; (ii) these cancers follow an autosomal dominant inheritance mechanism in which the neoplasm occurs in 1st degree relatives; and (iii) the formation of multiple tumors [3][4]. In the hereditary form of GI cancers, the first genetic mutation in one of the alleles of a predisposition gene is acquired at the time of conception, and the somatic mutation of the second allele is then acquired via environmental insult, lifestyle practices or other exogenous factors (Figure 1). Once the two alleles of a specific predisposition gene are mutated, gene function is completely inactivated, leading to carcinogenesis. Compared to the sporadic form of GI cancer, which requires two somatic events during the inactivation of the predisposition gene, hereditary cancers present a higher risk because they need only one somatic mutation event, which explains the early onset of hereditary cancers [5]. In parallel with the advancement of DNA technologies, the genetic mutations responsible for hereditary GI cancers have been widely documented (Table 1). These hereditary GI cancers include Cowden syndrome, MUTYH-associated polyposis, hereditary pancreatic cancer, Lynch syndrome, Peutz-Jeghers syndrome, familial adenomatous polyposis (FAP), attenuated FAP, serrated polyposis syndrome, and hereditary gastric cancer. Cancer-causing mutations can be initiated in three main classes of predisposition genes, oncogenes, tumor suppressor genes, and DNA repair genes, which are involved in establishing genetic stability.
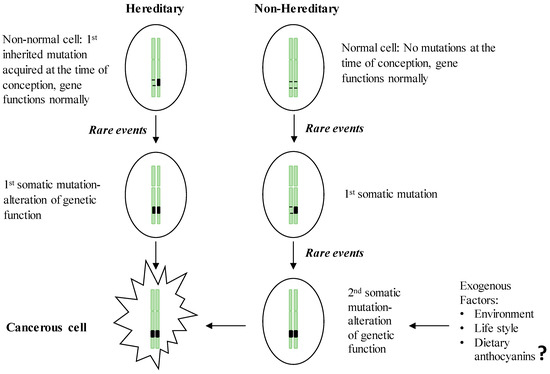
Figure 1. Two-hit theory of the initiation of hereditary and non-hereditary cancer. People with a hereditary susceptibility to GI cancers harbor an inherited genetic mutation on one of the chromosomes at the time of conception and receive the 1st somatic mutation due to the endogenous (e.g., chronic inflammation) or exogenous (e.g., exposure to carcinogens) rare events which in turn inactivate the full function of the respective gene and initiate neoplastic transformation. Non-inherited forms of GI cancer occur by acquiring two somatic mutations in later life, resulting in the inactivation of a gene leading to the initiation of malignancy.
Table 1. Hereditary basis of GI cancers.
| Type of the Cancer | Syndrome | Associated Germline Mutations | Reference | |
|---|---|---|---|---|
| Esophageal | Familial Barrett’s esophagus, Familial esophageal adenocarcinoma | MSR1, ASCC1 and CTHRC1 | [6] | |
| Tylosis with esophageal cancer-squamous cell carcinoma | RHBDF2 | [7] | ||
| Gastric | Diffuse hereditary gastric cancer-adenocarcinoma | CDH1 (E-cadherin) | [8] | |
| Pancreatic | Hereditary pancreatitis | PRSS1, CFTR, SPINK1, CTRC | [9] | |
| Hereditary breast and ovarian cancer | BRCA1/2 | |||
| Peutz-Jeghers syndrome | STK11/LKB1 | |||
| Familial atypical multiple mole melanoma syndrome |
CDKN2A/p16 | |||
| Familial adenomatous polyposis | APC | |||
| Colorectal | Familial adenomatous polyposis | APC | [10][11] | |
| Lynch syndrome | EPCAM, MLH1, MSH2, MSH6, PMS2 | |||
| MYH associated polyposis | MUTYH | |||
| Hamartomatous polyposis syndrome | Peutz-Jeghers syndrome | STK11 | ||
| Juvenile polyposis syndrome | SMAD4, BMPR1A | |||
| Attenuated Familial adenomatous polyposis | APC | |||
| Small intestine | Familial adenomatous polyposis | APC | [12] | |
| Lynch syndrome | Mutations in mismatch repair genes | |||
| Juvenile polyposis syndrome | SMAD4 | |||
| Peutz-Jeghers syndrome | STK11 | |||
| Liver | α-1 antitrypsin deficiency | SERPINA1 | [13][14][15][16][17] | |
| Hereditary hemochromatosis | HFE | |||
| Hereditary tyrosinemia type 1 | FAH | |||
| Glycogen storage disease type 1 | G6PC, SLC37A4 | |||
| Wilson’s disease | ATP7B | |||
| Niemann-park disease | SMPD1 AND NPC1 OR NPC2 | |||
| Biliary | Bile salt export pump deficiency | ABCB11 |
[18] |
|
2. Non-Hereditary GI Cancers
Accumulations of sporadic mutations can occur due to factors such as exposure to carcinogens [2], a westernized diet [19][20], diets rich in salt [21][22], obesity [23][24], chronic alcohol consumption [25][26], and chronic inflammation [27]. The relationships between carcinogens, diet, inflammation, and GI cancers are multiple and complex. Exposure to carcinogens can initiate cancer development via somatic mutations that include point mutations, deletions, additions, and modified methylation of DNA [28]. There are several cellular mechanisms to protect DNA from carcinogen-induced mutations and to identify and correct these mutations before they give rise to malignancy. In spite of these protective mechanisms, the GI tract is continuously exposed to chemical and biological carcinogens, often due to diets that act as carriers of preformed carcinogens [2]. Among known carcinogens, tobacco smoke hydrocarbons are one of the most potent, being comprised of more than 60 mutagens and cancer-causing chemicals directly linked to esophageal [28], pancreatic [29], and gastric cancers [30][31]. In addition, exposure to airborne occupational carcinogens such as cement dust, quartz dust, and diesel exhaust fumes increases the risk of gastric cancer [32]. Nitrosamines, which are produced from the chemical reaction between nitrates or, in reduced form, nitrites with amines present in meat products during the meat preservation process, are another group of potent carcinogens associated with the increased risk of malignancy in the liver and GI tract due to DNA alkylation and DNA adduct formation [33]. Among biological carcinogens, aflatoxin B1 is one of the most influential hepatocarcinogens produced by the Aspergillus flavis fungi. Due to its lipophilic nature, aflatoxin B1 is readily absorbed from the GI tract. Aflatoxin B1, upon its metabolism by cytochrome P450 in the liver, induces irreversible mutations in the p53 gene of hepatocytes [34][35]. Fumonisin B1 is another mycotoxin that can cause hepatic and esophageal cancers [36]. Fumonisin B1 acts in part by upregulating the production of inflammatory cytokines by gastric and colon epithelial cells [37].
GI tract carcinogenesis is attributed to chronic inflammatory conditions that occur due to microbial, viral, or disease conditions such as inflammatory bowel disease (IBD). Regarding microbial inflammation, Helicobacter pylori infection is well documented as a trigger for GI cancers. Exposure to H. pylori initiates active chronic gastritis by increasing the infiltration of inflammatory cells, which leads to the formation of intestinal adenocarcinoma and other malignant tumors of the GI tract [38]. In addition, hepatitis B and C virus infection, gastroesophageal reflux, enzyme damage, autoimmune diseases such as ulcerative colitis, and systemic stress conditions are responsible for chronic GI inflammation [2]. Clinical studies show that patients with IBD have a significantly higher risk of developing colorectal cancer, especially 8 to 10 years after the diagnosis of IBD [39]. The mechanisms underlining the link between inflammation and GI cancers are varied and include the production of high levels of reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI) [40]. The macrophages that dominate the chronic inflammatory microenvironment produce increased levels of ROS and RNI, which in turn interact with the DNA of proliferating epithelial cells and generate permanent genetic mutations leading to the malignant transformation. Excessive ROS/RNI production during the process of oxidative metabolism has been reported to promote the synthesis and secretion of inflammation-promoting cytokines such as tumor necrosis factor (TNF)-α, interferon-gamma (IFN-γ), and interleukin (IL)-6 [41][42]. Moreover, other inflammatory mediators such as chemokines, growth factors, and eicosanoids in tumor microenvironments contribute to inflammation-triggered tumor progression and metastasis via modulating the immune response, inhibiting apoptosis, inducing cell proliferation, and promoting the accumulation of oncogenic mutations [43].
Diet, personal lifestyle, and the environment are all linked to the development of GI cancers. Chronic excessive caloric intake and physical inactivity leading to overweight and obesity-derived metabolic dysfunction are essential risk factors of GI carcinogenesis [23][24]. Energy imbalance causes alterations in glycemic control, insulin signaling, and upregulation of adipose tissue-derived inflammatory pathways that prolong carcinogenesis-promoting conditions [44]. Chronic alcohol consumption also increases susceptibility to GI cancers [45]. Acetaldehyde, the primary metabolite of alcohol, has recently been targeted for its involvement in ethanol-linked oxidative stress and the inhibition of DNA methylation by interfering with the metabolism of B vitamins, reducing the activity of methionine synthase and glutathione levels [46], and disrupting retinoid metabolism [45]. Depletion of systemic and tissue-specific retinoic acid levels are associated with possible malignant transformation; thus, chronic alcohol consumption reduces hepatic vitamin A and retinoic acid levels, which are strongly related to the later development of hepatocellular carcinoma (HCC) via decreasing mitogen-activated protein kinase (MAPK) and increasing levels of phosphorylated c-Jun N-terminal kinases (JNKs) [45]. Moreover, in vivo evidence reveals that high-fat and high-salt diets alter the permeability and growth of the colonic mucus layer, which in turn, leads to intestinal microbial dysbiosis that is linked with an increased incidence of GI cancer [47]. Colonic microbial imbalance resulting from prolonged consumption of westernized diets enhances the breakdown and metabolism of specific glycans in the mucus layer, leading to GI carcinogenesis [48].
References
- Thirumurthi, S.; Vilar, E.; Lynch, P.J. Hereditary Gastrointestinal Cancers. In Textbook of Gastrointestinal Oncology; Springer: Cham, Switzerland, 2019; pp. 595–611.
- Katona, B.W.; Lynch, J.P. Mechanisms of Gastrointestinal Malignancies. In Physiology of the Gastrointestinal Tract: Sixth Edition; Elsevier Inc: Cambridge, MN, USA, 2018; Volume 2, pp. 1615–1642.
- Rahner, N.; Steinke, V. Hereditary cancer syndromes. Dtsch. Arztebl. 2008, 105, 706–714.
- Coleman, W.B. Neoplasia. In Molecular Pathology: The Molecular Basis of Human Disease; Elsevier Inc.: Cambridge, MN, USA, 2018; pp. 71–97.
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823.
- Van Nistelrooij, A.M.J.; Dinjens, W.N.M.; Wagner, A.; Spaander, M.C.W.; van Lanschot, J.J.B.; Wijnhoven, B.P.L. Hereditary factors in esophageal adenocarcinoma. Gastrointest. Tumors 2014, 1, 93–98.
- Ellis, A.; Risk, J.M.; Maruthappu, T.; Kelsell, D.P. Tylosis with oesophageal cancer: Diagnosis, management and molecular mechanisms. Orphanet. J. Rare Dis. 2015, 10, 126.
- Colvin, H.; Yamamoto, K.; Wada, N.; Mori, M. Hereditary gastric cancer syndromes. Surg. Oncol. Clin. N. Am. 2015, 24, 765–777.
- Eguchi, H.; Kobayashi, S.; Gotoh, K.; Noda, T.; Doki, Y. Characteristics of early-onset pancreatic cancer and its association with familial pancreatic cancer and hereditary pancreatic cancer syndromes. Ann. Gastroenterol. Surg. 2020, 4, 229–233.
- Wells, K.; Wise, P.E. Hereditary colorectal cancer syndromes. Surg. Clin. N. Am. 2017, 97, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Kanth, P.; Grimmett, J.; Champine, M.; Burt, R.; Samadder, J.N. Hereditary colorectal polyposis and cancer syndromes: A primer on diagnosis and management. Am. J. Gastroenterol. 2017, 112, 1509–1525.
- Shenoy, S. Genetic risks and familial associations of small bowel carcinoma. World J. Gastrointest. Oncol. 2016, 8, 509.
- Vanier, M.T. Niemann-Pick diseases. In Handbook of Clinical Neurology; Elsevier B.V.: Berlin/Heidelberg, Germany, 2013; Volume 113, pp. 1717–1721. [Google Scholar]
- Yang Chou, J.; Mansfield, B.C. Molecular genetics of type 1 glycogen storage diseases. Trends Endocrinol. Metab. 1999, 10, 104–113. [Google Scholar] [CrossRef]
- Villanueva, A.; Newell, P.; Hoshida, Y. Inherited hepatocellular carcinoma. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 725–734. [Google Scholar] [CrossRef]
- Chang, I.J.; Hahn, S.H. The genetics of Wilson disease. In Handbook of Clinical Neurology; Elsevier B.V.: Berlin/Heidelberg, Germany, 2017; Volume 142, pp. 19–34. [Google Scholar]
- MacIas, I.; Laín, A.; Bernardo-Seisdedos, G.; Gil, D.; Gonzalez, E.; Falcon-Perez, J.M.; Millet, O. Hereditary tyrosinemia type I-associated mutations in fumarylacetoacetate hydrolase reduce the enzyme stability and increase its aggregation rate. J. Biol. Chem. 2019, 294, 13051–13060.
- Gidi, A.D.G.; González-Chávez, M.A.; Villegas-Tovar, E.; Visag-Castillo, V.; Pantoja-Millan, J.P.; Vélez-Pérez, F.M.; Cano-García, F.; Contreras, A.G. An unusual type of biliar cyst: A case report. Ann. Hepatol. 2016, 15, 788–794.
- Moss, A.; Nalankilli, K. The association between diet and colorectal cancer risk: Moving beyond generalizations. Gastroenterology 2017, 152, 1821–1823. [Google Scholar] [CrossRef]
- Mehta, R.S.; Song, M.; Nishihara, R.; Drew, D.A.; Wu, K.; Qian, Z.R.; Fung, T.T.; Hamada, T.; Masugi, Y.; da Silva, A.; et al. Dietary patterns and risk of colorectal cancer: Analysis by tumor location and molecular subtypes. Gastroenterology 2017, 152, 1944–1953.
- Gaddy, J.A.; Radin, J.N.; Loh, J.T.; Zhang, F.; Kay Washington, M.; Peek, R.M.; Scott Algood, H.M.; Cover, T.L. Hiharriscagh dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect. Immun. 2013, 81, 2258–2267. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, L.; Galletti, F.; Strazzullo, P. Dietary salt intake and risk of gastric cancer. Cancer Treat. Res. 2014, 159, 83–95.
- Ulrich, C.M.; Himbert, C.; Holowatyj, A.N.; Hursting, S.D. Energy balance and gastrointestinal cancer: Risk, interventions, outcomes and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 683–698. [Google Scholar] [CrossRef]
- Gunter, M.J.; Riboli, E. Obesity and gastrointestinal cancers—Where do we go from here? Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 651–652.
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293.
- Sierra, J.C.; Piazuelo, M.B.; Luis, P.B.; Barry, D.P.; Allaman, M.M.; Asim, M.; Sebrell, T.A.; Finley, J.L.; Rose, K.L.; Hill, S.; et al. Spermine oxidase mediates Helicobacter pylori-induced gastric inflammation, DNA damage, and carcinogenic signaling. Oncogene 2020, 1–10.
- Huang, F.L.; Yu, S.J. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J. Surg. 2018, 41, 210–215.
- Bao, Y.; Giovannucci, E.; Fuchs, C.S.; Michaud, D.S. Passive smoking and pancreatic cancer in women: A prospective cohort study. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2292–2296.
- Praud, D.; Rota, M.; Pelucchi, C.; Bertuccio, P.; Rosso, T.; Galeone, C.; Zhang, Z.F.; Matsuo, K.; Ito, H.; Hu, J.; et al. Cigarette smoking and gastric cancer in the Stomach Cancer Pooling (StoP) Project. Eur. J. Cancer Prev. 2018, 27, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Moy, K.A.; Fan, Y.; Wang, R.; Gao, Y.T.; Yu, M.C.; Yuan, J.M. Alcohol and tobacco use in relation to gastric cancer: A prospective study of men in Shanghai, China. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2287–2297.
- Sjödahl, K.; Jansson, C.; Bergdahl, I.A.; Adami, J.; Boffetta, P.; Lagergren, J. Airborne exposures and risk of gastric cancer: A prospective cohort study. Int. J. Cancer. 2007, 120, 2013–2018.
- De Oliveira, G.A.; Cheng, R.Y.S.; Ridnour, L.A.; Basudhar, D.; Somasundaram, V.; McVicar, D.W.; Monteiro, H.P.; Wink, D.A. Inducible nitric oxide synthase in the carcinogenesis of gastrointestinal cancers. Antioxid. Redox Signal. 2017, 26, 1059–1077.
- Adam, M.A.A.; Tabana, Y.M.; Musa, K.B.; Sandai, D.A. Effects of different mycotoxins on humans, cell genome and their involvement in cancer (Review). Oncol. Rep. 2017, 37, 1321–1336. [Google Scholar] [CrossRef]
- Marchese, S.; Polo, A.; Ariano, A.; Velotto, S.; Costantini, S.; Severino, L. Aflatoxin B1 and M1: Biological properties and their involvement in cancer development. Toxins 2018, 10, 214.
- Myburg, R.B.; Dutton, M.F.; Chuturgoon, A.A. Cytotoxicity of fumonisin B1, diethylnitrosamine, and catechol on the SNO esophageal cancer cell line. Environ. Health Perspect. 2002, 110, 813–815.
- Mahmoodi, M.; Alizadeh, A.M.; Sohanaki, H.; Rezaei, N.; Amini-Najafi, F.; Khosravi, A.R.; Hosseini, S.K.; Safari, Z.; Hydarnasab, D.; Khori, V. Impact of fumonisin B1 on the production of inflammatory cytokines by gastric and colon cell lines. Iran. J. Allergy Asthma Immunol. 2012, 11, 165–173.
- Alfarouk, K.O.; Bashir, A.H.H.; Aljarbou, A.N.; Ramadan, A.M.; Muddathir, A.K.; AlHoufie, S.T.S.; Hifny, A.; Elhassan, G.O.; Ibrahim, M.E.; Alqahtani, S.S.; et al. The possible role of Helicobacter pylori gastric cancer and its management. Front. Oncol. 2019, 9, 75.
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801.
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121.
- Vaidya, F.U.; Chhipa, A.S.; Sagar, N.; Pathak, C. Oxidative Stress and Inflammation Can Fuel Cancer. In Role of Oxidative Stress in Pathophysiology of Diseases; Springer: Singapore, 2020; pp. 229–258. [Google Scholar]
- Yu, J.H.; Kim, H. Oxidative stress and cytokines in the pathogenesis of pancreatic cancer. J. Cancer Prev. 2014, 19, 97–102.
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386.
- O’Sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714.
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612.
- Stickel, F.; Schuppan, D.; Hahn, E.G.; Seitz, H.K. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut 2002, 51, 132–139.
- Schroeder, B.O.; Birchenough, G.M.H.; Ståhlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Bäckhed, F. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe 2018, 23, 27–40.
- Weng, M.T.; Chiu, Y.T.; Wei, P.Y.; Chiang, C.W.; Fang, H.L.; Wei, S.C. Microbiota and gastrointestinal cancer. J. Formos. Med. Assoc. 2019, 118, 32–41.


