Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Visentin | + 2588 word(s) | 2588 | 2022-03-21 05:13:27 | | | |
| 2 | Amina Yu | + 6 word(s) | 2594 | 2022-03-21 09:34:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Visentin, A. Monoclonal Gammopathies of Neurological Significance. Encyclopedia. Available online: https://encyclopedia.pub/entry/20790 (accessed on 01 August 2026).
Visentin A. Monoclonal Gammopathies of Neurological Significance. Encyclopedia. Available at: https://encyclopedia.pub/entry/20790. Accessed August 01, 2026.
Visentin, Andrea. "Monoclonal Gammopathies of Neurological Significance" Encyclopedia, https://encyclopedia.pub/entry/20790 (accessed August 01, 2026).
Visentin, A. (2022, March 21). Monoclonal Gammopathies of Neurological Significance. In Encyclopedia. https://encyclopedia.pub/entry/20790
Visentin, Andrea. "Monoclonal Gammopathies of Neurological Significance." Encyclopedia. Web. 21 March, 2022.
Copy Citation
Monoclonal gammopathies of neurological significance include a widespread range of manifestations, ranging from slowly progressive sensitive demyelinating polyneuropathy with anti-MAG antibody to subacute rapidly progressive forms as in POEMS syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammopathy, Skin changes) or neurolymphomatosis.
monoclonal gammopathies of neurological significance (MGNS)
anti-myelin-associated-glycoprotein (MAG) polyneuropathy
POEMS syndrome
ibrutinib
rituximab
venetoclax
1. Signaling Pathways in B-Cell Malignancies
The B-cell receptor (BCR), the toll-like receptors (TLR), and the C-X-C chemokine receptor type 4 (CXCR4) pathways are key signaling pathways which tune the survival, proliferation, activation, migration and metabolism of B-cell biology.
The BCR is a major actor in the regulation of B-cells biology, as well as in the oncogenesis of B-cell-derived NHL (Figure 1). After antigen binding to the BCR variable regions, the Bruton’s tyrosine kinase (BTK) is activated, promoting the proteasomal degradation of IkB (inhibitor of NF-kB), which subsequently modulates NF-kB activation and downstream proteins [1]. For this reason, ibrutinib, the first in class covalent BTK inhibitor, but also acalabrutinib and zanubrutinib, showed prolonged responses within clinical trials and became the standard of care for most chronic B-cell malignancies. The sustainment of the neoplastic lymphocytes is not only favored by activation of survival and proliferation signaling pathways, but also by disruption of anti-apoptotic proteins. BCL2 (B-cell lymphoma 2) protein is the leading anti-apoptotic protein regulating the BAX-BAK mitochondria channel, the release of cytochrome C and the activation of the intrinsic pathway of apoptosis [2]. BCL2 is overexpressed in several hematological malignancies such as chronic lymphocytic leukemia (CLL) and aggressive NHL, and is targeted by venetoclax [2]. Venetoclax acts binding BCL-2, displacing Bim and other BH-3 only proteins from BLC2. Bim can bind to BAX-BAK, favoring the activation of apotosis [2].
After binding of pathogen-associated molecular patterns (PAMP) to the TLR and recruitment of myeloid differentiation primary response 88 (MYD88) and IRAKs forming the “myddosome complex”, the transcriptional factor NF-kB is activated (Figure 1). Ngo and colleagues identified for the first time somatic activating mutations in MYD88, leading to change of the 265th amino-acid MYD88 (MYD88L265P) from leucine to proline [3]. Several other studies confirmed recurrent MYD88L265P mutation in B-cell NHL, by using conventional Sanger sequencing, allele-specific polymerase chain reaction (PCR) analysis, but also NGS technologies on bone marrow cells [3][4]. The MYD88L265P mutation was found in 90% of patients with IgM Waldenstron’s macroglobulinemia (WM)/lymphoplasmocytic lymphoma (LPL), in 55% of non-IgM LPL (55%) and almost half of IgM monoclonal gammopathies of undetermined significance (MGUS) [5]. In central nervous system lymphomas, orbital/vitreoretinal lymphomas, intravascular large B-cell lymphomas, the prevalence of MYD88L265P ranges from 44% to 73% [4][5]. Conversely, MYD88L265P is rarely present in splenic marginal zone lymphoma (7.0%), CLL (2.5%), multiple myeloma (1.5%), and is usually absent in IgG or IgA MGUS [4][5]. Other somatic mutations in the MYD88 gene have been identified, but their impact is largely unknown [6]. The MYD88L265P mutation favors the assemble of the complex, boosting the activation downstream signaling [3].
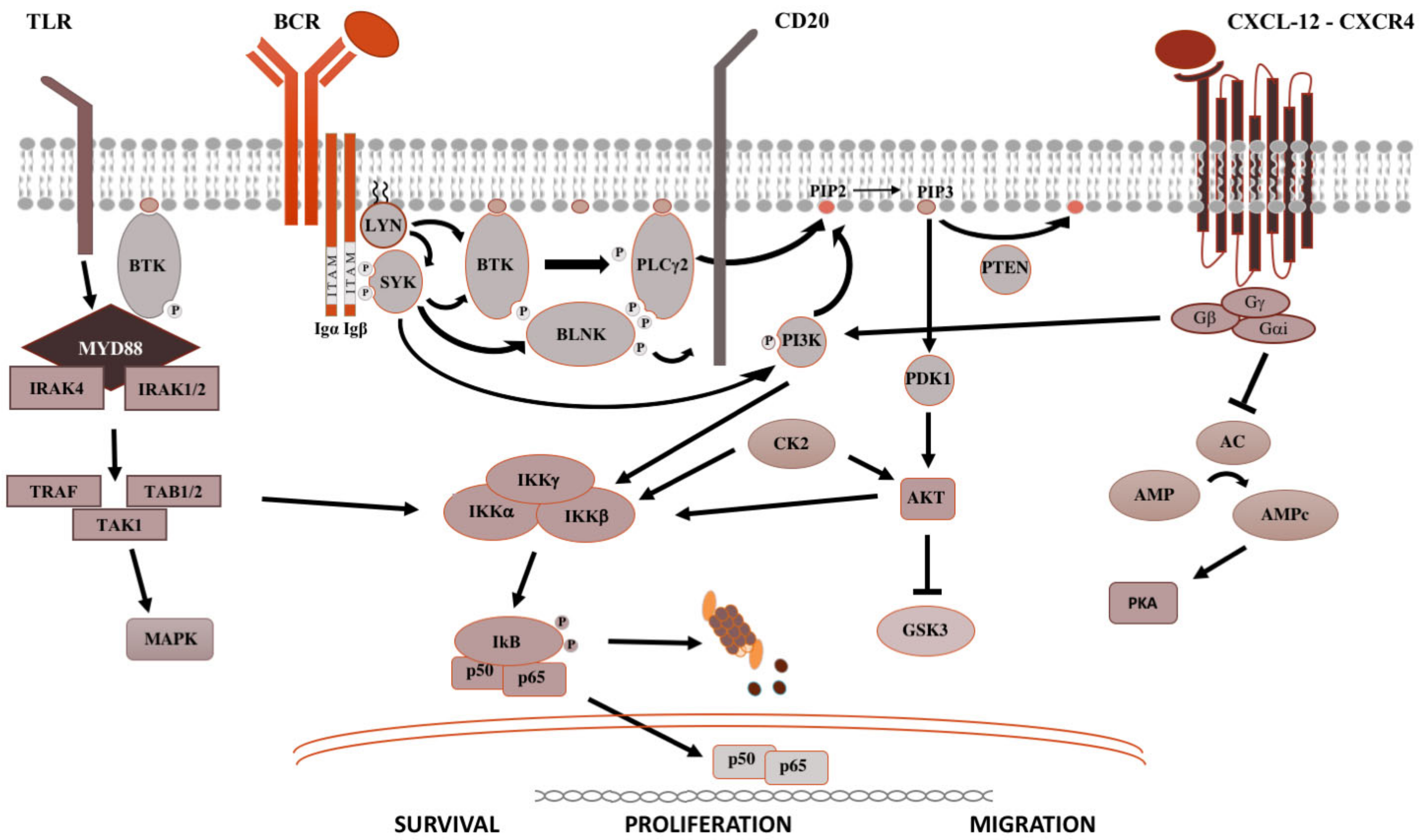
Figure 1. Pivotal signaling pathways in lymphoproliferative diseases. On the left side, TLR signaling is shown. Upon recognition of PAMP (pathogen-associated molecular patterns) by TLR (toll-like receptors), MYD88 is recruited to the cell membrane, binds the serine-threonine kinase IRAK proteins (IL-1R associated kinase), TRAF (tumor necrosis factor receptor-associated factor) and TAK1 (transforming growth factor beta-activated kinase 1) [7], which activate the MAPK (mitogen-activated protein kinase) cascade and IKK (inhibitor of the NF-κB kinase). IKK complex is made up of IKKα and IKKβ, with kinasic activity, and the regulatory IKKγ subunit. Once activated, IKK phosphorylates IκB (inhibitor of NF-κB), leading to its ubiquitylation and proteasomal degradation, thereby, the release of the NF-κB (RELA p65–p50 in the classical pathway and RELB-p52 in the alternative pathway). Then, NF-κB subunits translocate into the nucleus and modulate the proliferation and survival of B lymphocytes [1]. In the middle is shown the BCR signaling. After antigen binding to the B-cell receptor (BCR), the Src kinase LYN is activated, phosphorylating CD79A/B and recruiting SYK (spleen tyrosine kinase) [8]. LYN and SYK activate by phosphorylation BTK [9], and, subsequently, the phospholipase PLCγ2, activating NF-kB pathway, MAPK and PI3K pathways. On the right is shown the CXCR4 signaling. After binding of CXCL12 to the seven domains trans-membrane CXCR4 receptor coupled with trimeric G protein, the Gα subunit dissociates from the Gβ/Gγ dimer and inhibits adenyl cyclase (AC), causing the reduction of intracellular cAMP (cyclic AMP) levels and switching off PKA (protein kinase A). Meanwhile, the Gβ/Gγ dimer actives PI3K, modulating lymphocytes migration, and phospholipase PLC, which acts on PIP2 (phosphatidylinositol 4,5-bisphosphate) releasing IP3 (inositol 1,4,5-trisphosphate), favoring the mobilization of Ca2+ from intracellular stores, and diacylglycerol, promoting the activation of PKC (protein kinase C) and MAPK cascade [1].
CXCR4 is one of the most important receptors involved in the regulation of homing and egress of hematological cells trafficking from bone marrow blood and secondary lymphoid organs (Figure 1) [10]. Acquired mutations of CXCR4 gene, such as R334X, G336X, S338X (the most common), E343X, have been described in up to 40%, causing the truncation of the C-terminal tail of this transmembrane protein [11].
Both MYD88 and CXCR4 mutations should be evaluated in bone marrow cells, according to hematological guidelines [11]. However, some findings suggest that MYD88L265P mutation may be evaluated in the peripheral blood cell or by using cell free DNA, thus avoiding invasive diagnostic procedures [12][13]. On the other hand, bone marrow biopsy is recommended in order the correctly diagnose patients’ IgM gammopathy [11].
2. Monoclonal Gammopathies of Neurological Significance
The association between peripheral neuropathy and monoclonal protein has been extensively investigated in the literature, being first reported of myeloma and WM [14]. However, the exact prevalence of monoclonal gammopathy of neurological significance is unknown and likely underrated. Monoclonal proteins have been found to be present in 10% of otherwise unexplained peripheral neuropathies [15], with a frequency 6 to 10 times greater than in the general population. The occurrence of both paraprotein and neuropathy increases with age [16] and the association might be casual. Whereas, the most common paraprotein is the IgG isotype, neuropathy, usually demyelinating, is much more common in patients carrying an IgM paraprotein (50–75%, compared with 17–35% with IgG and 8–15% with IgA) [14]. When an association between the hematological diseases and the polyneuropathy is suspected, due to the high prevalence of MGUS in the general population, the first step is to exclude common causes of secondary polyneuropathy such as diabetes mellitus, vitamin deficiency, chronic alcohol consumption, drugs, HCV infection, and so on. For these reasons, a multidisciplinary assessment involving hematologists, neurologists, radiologists and neurophysiologists should be guided based on the type of the paraprotein IgM vs. IgG/IgA. In selected cases, nerve biopsy (including light microscopic examination, immunocytochemical techniques and electron microscopic imaging) may be necessary, especially when suspecting vasculitis or neurolymphomatosis (see below). Summary flow charts for IgM and IgG/IgA monoclonal-gammopathy-related peripheral neuropathies are shown in Figure 1 and Figure 2.
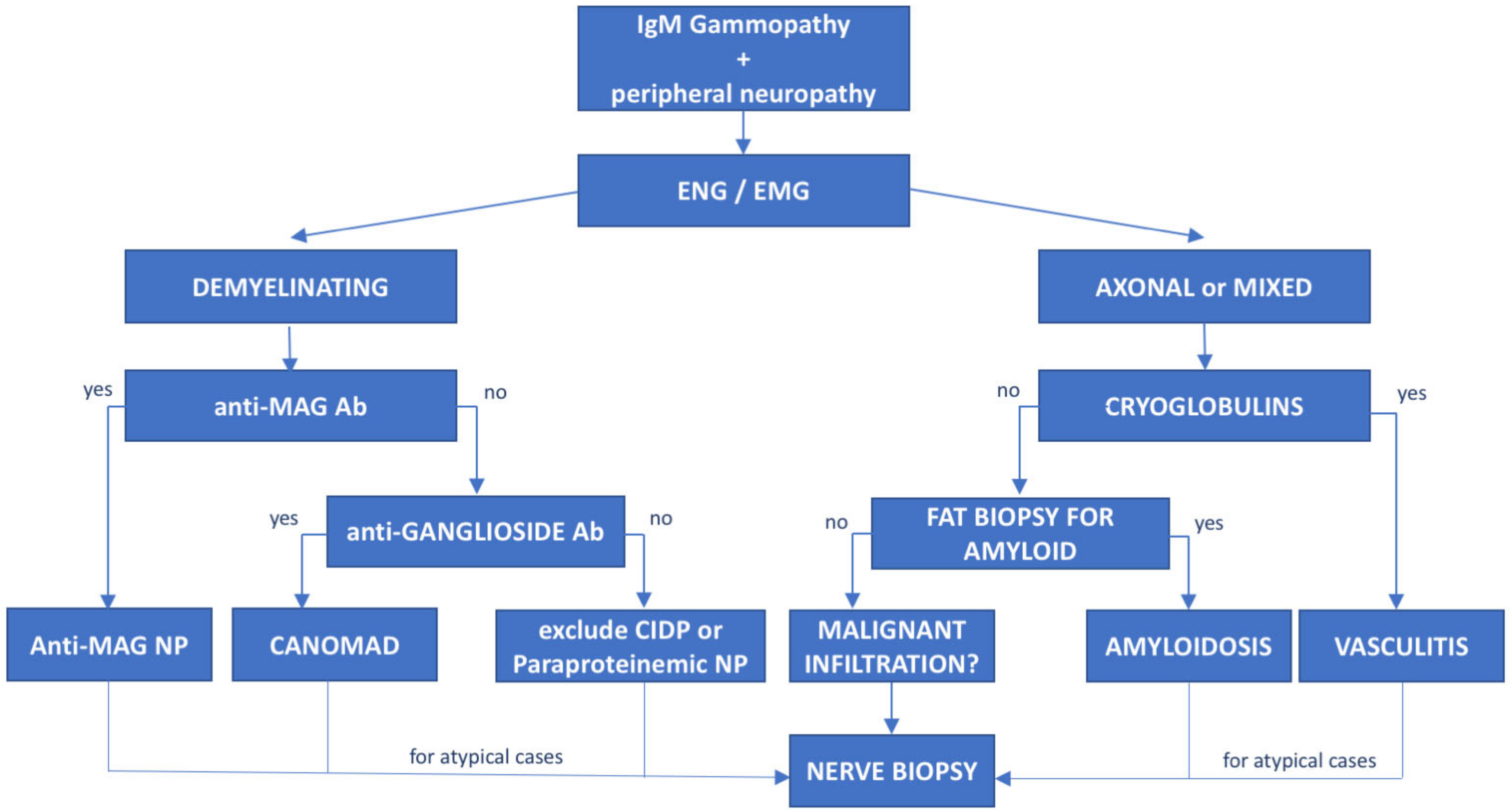
Figure 2. Diagnostic flowchart of peripheral neuropathy associated with IgM monoclonal gammopathy. Patients with IgM monoclonal protein and peripheral neuropathy diagnosed with neurophysiological tests (electroneurography (ENG) and electromyography (EMG)) should undergo further investigation based on the type of neuropathy. Demyelinating cases with prolonged distal latency must be tested for anti-MAG antibodies to rule out anti-MAG neuropathy; negative cases with chronic ataxia and ophthalmoplegia must be tested for antiganglioside antibodies to exclude CANOMAD diseases. In double-negative patients, CIDP and paraproteinemic neuropathy should be taken into consideration. On the other hand, for patients with axonal or mixed (axonal and demyelinating features) neuropathies, cryoglobulins and light-chain amyloid deposition in fat biopsy should be investigated. However, for atypical or rapidly progressive cases, nerve biopsy is recommended to exclude neurolymphomatosis. Ab—antibodies, NP—neuropathy.
3. Monoclonal Gammopathies of Undetermined Significance
MGUS is a preneoplastic condition which can be found in approximately 3% of people ≥50 years [17], diagnosed by the presence of (i) monoclonal protein <30 g/L, (ii) <10% clonal neoplastic cells within the bone marrow, (iii) lack of organomegaly and end-organ damage, according to the Mayo clinic guidelines. MGUS can be caused by the presence of a full antibody (heavy-chain and light-chain), further classified according to the type of the heavy chain in IgG, IgM, IgA and IgD MGUS, and/or light-chain-only.
It can progress to overt neoplasia and is associated with shorter survival than age-matched control population [17]. While almost all patients with multiple myeloma and WM usually had an MGUS phase, not all people with MGUS will develop a hematological disease. The most common MGUS subtype is IgG MGUS, but it is associated with a low risk of developing multiple myeloma—1% people/year if monoclonal protein is <15 g/L and the free–light chain ratio is within normal range [17]. Conversely, IgM MGUS harbors the highest risk of evolution to indolent lymphoma such as WM, marginal zone lymphoma or CLL (described afterwards) [17].
The knowledge of the genomic profiling of IgM gammopathies has favored the achievement of significant therapeutic improvement. Gain-of-function L265P mutation of MYD88, the most commonly mutated gene in WM and IgM-MGUS [4], favors the activation of BTK and of NF-κB signaling (Figure 1). The CXCR4 gene is less commonly mutated, usually by nonsense or frameshift mutations, but it has been shown to be associated with lower response rates and early relapse [11] (Figure 1). Recently, Varettoni et al. [18] found that MYD88-mutated patients with at least 10 g/L of paraprotein have the highest risk of progression to WM or other lymphoproliferative disease (11.6% at 5 years and 38% at 10 years from the IgM MGUS diagnosis).
The IgM reacts against myelin-associated glycoprotein (MAG) [19][20]. Anti-MAG neuropathy is described below. IgM paraproteinemic neuropathy may present with antibodies to peripheral nerve antigens different from MAG [21], or it may not present antibody reactivity at all [22][23]. In such cases, also with a rapid neurological progression, pain, early prominent motor involvement or axonal damage, AL amyloidosis or cryoglobulinemic vasculitis should be considered. Sometimes, in patients with a multifocal motor neuropathy with conduction block phenotype, an IgM paraprotein with antiganglioside GM1 antibodies can also be detected [14]. Bing–Neel syndrome is a rare complication of WM, caused by the infiltration of neoplastic B-cells in the cerebral parenchyma, proximal nerve roots, peripheral nerves and meninges. Recurrent manifestations include headaches, cognitive and psychiatric dysfunction, seizures, cranial and peripheral neuropathies. MYD88L265P can be found in the cerebral spinal fluid of almost all patients [24]. Despite this aggressive presentation, Bing–Neel syndrome responds well to BTK inhibitors [25].
4. Waldenström’s Macroglobulinemia/Lymphoplasmacytic Lymphoma
Waldenström’s macroglobulinemia is characterized by the proliferation of mature lymphoplasmacytic B-cells which release macroglobulin, most commonly an IgM paraprotein or more rarely an IgA or IgG subtype. While MYD88L265P mutation is found in almost all patients with IgM-WM, CXCR4WHIM-like mutation is found in less than one-third of patients, usually at subclonal level, and is associated with a larger disease burden, lower response to ibrutinib, and usually decreased overall survival due to early relapse [4][11].
Peripheral axonal and/or demyelinating neuropathies are common in Waldenstron’s macroglobulinemia (WM), being found up to 62.5% of patients, with different manifestations depending on the underlying pathogenic mechanism. Demyelinating polyneuropathy in WM is commonly symmetric, distal, slowly progressive and associated with IgM antibodies against nerve antigens [21][26], most commonly MAG. The presence of polyneuropathy is a criterion for WM treatment. Patients with MW may also develop axonal polyneuropathies caused by drugs such as bortezomib, cryoglobulins, light-chain amyloid deposition, B-cell infiltration, and rare CANOMAD disease characterized by chronic ataxic neuropathy, ophthalmoplegia, IgM M-protein, positivity for cold agglutinins and antibodies to disialosyl antigens (GD1b, GQ1b, GT1a, GT1b) [27].
Cryoglobulinemias can be classified as follows: type I—monoclonal immunoglobulin (more common IgM > IgG > IgA); type II—monoclonal IgM and polyclonal IgG; type III—polyclonal IgM with rheumatoid factor activity and polyclonal IgG. While type II is more common in WM, type III is seen predominantly in connective tissue diseases or in chronic infections such as chronic HCV hepatitis [28]. Monoclonal IgMs cryoglobulins may precipitate below 37 °C, causing severe painful multifocal neuropathy, sometimes involving cranial nerves. Patients can also suffer from arthralgia, glomerulonephritis, skin ulceration or purpura.
5. Anti-Myelin-Associated Glycoprotein (MAG) Antibody Neuropathy
Polyneuropathy with IgM anti-MAG antibodies and IgM MGUS, but also WM, marginal zone lymphoma (MZL) or CLL [29] is the most common IgM paraproteinemic polyneuropathy [30]. Anti-MAG antibodies have a pathogenic role, as demonstrated by experimental mice models and human sural nerve biopsies, showing IgM and complement deposition, as well as characteristic widening of myelin lamellae on electron microscopy investigation leading to the segmentation of myelin [29].
Patients usually present with a length-dependent slowly progressive symmetric polyneuropathy, with predominant sensory impairment at lower limbs, resulting in sensory ataxia, and tremor at upper limbs, with a significant impact on patients’ disability and quality of life [31]. Motor involvement generally occurs late in the course of the disease. Regarding neurophysiological features, a demyelinating neuropathy with symmetric reduction of conduction velocities and sensory nerve action potentials is observed with the typical finding of disproportionately prolonged distal motor latencies and lack of conduction blocks. Axonal features may appear in addition to demyelination in patients with a long disease duration.
To date, inadequate evidence has been obtained from therapeutic trials [32]. After initial results from small studies, rituximab, an anti-CD20 chimeric monoclonal antibody, was evaluated in two randomized clinical trials with controversial results [33][34]. Remarkably, patients with WM were excluded from these trials. Real-life retrospective studies agree that rituximab is active in almost 30–50% of the patients [33][34], with the same rate of responses in patients with WM or IgM MGUS [35]. Albeit with low-quality evidence, a Cochrane meta-analysis [32] confirmed that rituximab improves disability scales and the response to questionnaires in the global impression of anti-MAG antibody neuropathy. Therefore, rituximab is currently used in the clinical practice either alone or in combination with cyclophosphamide [36], fludarabine [37] or bendamustine [38]. Obinutuzumab, a humanized glycoengineered anti-CD20 monoclonal antibody, has also been used as a possible alternative treatment in patients with anti-MAG antibody neuropathy, with controversial results and concerns regarding possible toxicity [39][40].
Recently, ibrutinib has proven to be active in 3 MYD88L265P-mutated and CXCR4 wild-type patients with anti-MAG antibody polyneuropathy and WM. Two of the three cases were also refractory to rituximab [41]. In particular, decrease of INCAT (Inflammatory Neuropathy Cause and Treatment) Disability and INCAT Sensory Sum (ISS) scores confirmed a neurological improvement. After 12 months of treatment, ibrutinib was safe in these elderly patients, and no events of atrial fibrillation or infections were recorded. In addition, second-generation BTK inhibitors, such as acalabrutinib [42] and zanubrutinib [43], showed promising results as single agents or in combination with rituximab for the treatment of symptomatic WM. These new drugs selectively target BTK and are associated with lower adverse events than ibrutinib [44]. Zanubrutinib displayed also encouraging results in MYD88 wild-type patients [45]. Venetoclax is an oral and selective BCL2 inhibitor that in combination with rituximab proved to be highly active in B-cell malignancies even after ibrutinib failure [46]. Recently, Castillo et al. showed that venetoclax was able to induce remission in relapsed WM regardless of CXCR4 mutations [47].
6. Immunoglobulin Light-Chain (AL) Amyloidosis
AL amyloidosis is caused by the misfolding and deposition of immunoglobulin light chains in tissues and organs released by lymphoplasmacytic cells in patients with MGUS, multiple myeloma or, less commonly, WM or CLL. Patients with AL amyloid neuropathy may display neuropathic pain and autonomic symptoms with orthostatic hypotension, diarrhea or constipation, pupil asymmetry, genitourinary and sexual dysfunction. In the diagnostic workup, it is useful to look for damages to heart (heart failure with septum thickness and preserved ejection fraction), kidney (albuminuria) and intrahepatic cholestasis (increase of bilirubin and alkaline phosphatase). Additional red flags in the diagnosis of AL amyloidosis are coagulation factor X deficiency, highly specific to AL amyloidosis but uncommon, sometimes favoring the development of “racoon eyes” (for example, periorbital ecchymoses) [48], and macroglossia. All these points make AL amyloidosis diagnostic delay quite common [48].
References
- Piazza, F.; Manni, S.; Arjomand, A.; Visentin, A.; Trentin, L.; Semenzato, G. New responsibilities for aged kinases in B-lymphomas. Hematol. Oncol. 2020, 38, 3–11.
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64.
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119.
- De Groen, R.A.L.; Schrader, A.M.R.; Kersten, M.J.; Pals, S.T.; Vermaat, J.S.P. MYD88 in the driver’s seat of B-cell lymphomagenesis: From molecular mechanisms to clinical implications. Haematologica 2019, 104, 2337–2348.
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462.
- Dubois, S.; Viailly, P.-J.; Bohers, E.; Bertrand, P.; Ruminy, P.; Marchand, V.; Maingonnat, C.; Mareschal, S.; Picquenot, J.-M.; Penther, D.; et al. Biological and Clinical Relevance of Associated Genomic Alterations in MYD88 L265P and non-L265P-Mutated Diffuse Large B-Cell Lymphoma: Analysis of 361 Cases. Clin. Cancer Res. 2017, 23, 2232–2244.
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890.
- Severin, F.; Frezzato, F.; Visentin, A.; Martini, V.; Trimarco, V.; Carraro, S.; Tibaldi, E.; Brunati, A.M.; Piazza, F.; Semenzato, G.; et al. In Chronic Lymphocytic Leukemia the JAK2/STAT3 Pathway Is Constitutively Activated and Its Inhibition Leads to CLL Cell Death Unaffected by the Protective Bone Marrow Microenvironment. Cancers 2019, 11, 1939.
- Frezzato, F.; Raggi, F.; Martini, V.; Severin, F.; Trimarco, V.; Visentin, A.; Scomazzon, E.; Accordi, B.; Bresolin, S.; Piazza, F.; et al. HSP70/HSF1 axis, regulated via a PI3K/AKT pathway, is a druggable target in chronic lymphocytic leukemia. Int. J. Cancer 2019, 145, 3089–3100.
- Martini, V.; Gattazzo, C.; Frezzato, F.; Trimarco, V.; Pizzi, M.; Chiodin, G.; Severin, F.; Scomazzon, E.; Guzzardo, V.; Saraggi, D.; et al. Cortactin, a Lyn substrate, is a checkpoint molecule at the intersection of BCR and CXCR4 signalling pathway in chronic lymphocytic leukaemia cells. Br. J. Haematol. 2017, 178, 81–93.
- Treon, S.P.; Xu, L.; Liu, X.; Hunter, Z.R.; Yang, G.; Castillo, J.J. Genomic Landscape of Waldenström Macroglobulinemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 745–752.
- Nakamura, A.; Ohwada, C.; Takeuchi, M.; Takeda, Y.; Tsukamoto, S.; Mimura, N.; Nagisa, O.-H.; Sugita, Y.; Tanaka, H.; Wakita, H.; et al. Detection of MYD88 L265P mutation by next-generation deep sequencing in peripheral blood mononuclear cells of Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. PLoS ONE 2019, 14, e0221941.
- Bagratuni, T.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Mavrianou-Koutsoukou, N.; Liacos, C.; Patseas, D.; Kanellias, N.; Migkou, M.; Ziogas, D.C.; Eleutherakis-Papaiakovou, E.; et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia 2018, 32, 2617–2625.
- Carroll, A.S.; Lunn, M.P.T. Paraproteinaemic neuropathy: MGUS and beyond. Pract. Neurol. 2021, 21, 492–503.
- Kelly, J.J., Jr.; Kyle, R.A.; O’Brien, P.C.; Dyck, P.J. Prevalence of monoclonal protein in peripheral neuropathy. Neurology 1981, 31, 1480–1483.
- Hanewinckel, R.; Drenthen, J.; van Oijen, M.; Hofman, A.; van Doorn, P.A.; Ikram, M.A. Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016, 87, 1892–1898.
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249.
- Varettoni, M.; Zibellini, S.; Boveri, E.; Klersy, C.; Candido, C.; Rattotti, S.; Ferretti, V.V.; DeFrancesco, I.; Mangiacavalli, S.; Nizzoli, M.E.; et al. A risk-stratification model based on the initial concentration of the serum monoclonal protein and MYD 88 mutation status identifies a subset of patients with IgM monoclonal gammopathy of undetermined significance at high risk of progression to Waldenström macroglobulinaemia or other lymphoproliferative disorders. Br. J. Haematol. 2019, 187, 441–446.
- Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. J. Neuroimmunol. 2021, 361, 577725.
- Latov, N. Antibody testing in neuropathy associated with anti-Myelin-Associated Glycoprotein antibodies: Where we are after 40 years. Curr. Opin. Neurol. 2021, 34, 625–630.
- Boso, F.; Ruggero, S.; Giannotta, C.; Benedetti, L.; Marfia, G.A.; Ermani, M.; Campagnolo, M.; Salvalaggio, A.; Gallia, F.; De Michelis, C.; et al. Anti-sulfatide/galactocerebroside antibodies in immunoglobulin M paraproteinemic neuropathies. Eur. J. Neurol. 2017, 24, 1334–1340.
- Niermeijer, J.; Fischer, K.; Eurelings, M.; Franssen, H.; Wokke, J.H.; Notermans, N.C. Prognosis of polyneuropathy due to IgM monoclonal gammopathy: A prospective cohort study. Neurology 2010, 74, 406–412.
- LaRue, S.; Bombelli, F.; Viala, K.; Neil, J.; Maisonobe, T.; Bouche, P.; Musset, L.; Fournier, E.; Léger, J.M. Non-anti-MAG DADS neuropathy as a variant of CIDP: Clinical, electrophysiological, laboratory features and response to treatment in 10 cases. Eur. J. Neurol. 2011, 18, 899–905.
- Frustaci, A.M.; Rusconi, C.; Picardi, P.; Veronese, S.M.; Montillo, M.; Cairoli, R.; Tedeschi, A. Bing Neel Syndrome in a Previously Untreated Patient with Waldenström’s Macroglobulinemia: Contribution of MYD88 L265P Mutation on Cerebrospinal Fluid. Clin. Lymphoma Myeloma Leuk. 2016, 16, e7–e9.
- Castillo, J.J.; Itchaki, G.; Paludo, J.; Varettoni, M.; Buske, C.; Eyre, T.A.; Chavez, J.C.; Shain, K.H.; Issa, S.; Palomba, M.L.; et al. Ibrutinib for the treatment of Bing-Neel syndrome: A multicenter study. Blood 2019, 133, 299–305.
- Campagnolo, M.; Ferrari, S.; Dalla Torre, C.; Cabrini, I.; Cacciavillani, M.; Lucchetta, M.; Ruggero, S.; Toffanin, E.; Cavallaro, T.; Briani, C. Polyneuropathy with anti-sulfatide and anti-MAG antibodies: Clinical, neurophysiological, pathological features and response to treatment. J. Neuroimmunol. 2015, 281, 1–4.
- Le Cann, M.; Bouhour, F.; Viala, K.; Simon, L.; Tard, C.; Rossi, C.; Morel, G.; Lagrange, E.; Magy, L.; Créange, A.; et al. CANOMAD: A neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood 2020, 136, 2428–2436.
- Muchtar, E.; Magen, H.; Gertz, M.A. How I treat cryoglobulinemia. Blood 2017, 129, 289–298.
- Briani, C.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Ferrari, S.; Cavallaro, T.; Manara, R.; Gasparotti, R.; Piazza, F. Peripheral nervous system involvement in lymphomas. J. Peripher. Nerv. Syst. 2019, 24, 5–18.
- Latov, N. Diagnosis and treatment of chronic acquired demyelinating polyneuropathies. Nat. Rev. Neurol. 2014, 10, 435–446.
- Campagnolo, M.; Ruiz, M.; Falzone, Y.M.; Ermani, M.; Bianco, M.; Martinelli, D.; Cerri, F.; Quattrini, A.; Salvalaggio, A.; Castellani, F.; et al. Limitations in daily activities and general perception of quality of life: Long term follow-up in patients with anti-myelin-glycoprotein antibody polyneuropathy. J. Peripher. Nerv. Syst. 2019, 24, 276–282.
- Lunn, M.P.; Nobile-Orazio, E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst. Rev. 2010, 2016, CD002827.
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293.
- Léger, J.-M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.-M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225.
- Campagnolo, M.; Zambello, R.; Nobile-Orazio, E.; Benedetti, L.; Marfia, G.A.; Riva, N.; Castellani, F.; Bianco, M.; Salvalaggio, A.; Garnero, M.; et al. IgM MGUS and Waldenstrom-associated anti-MAG neuropathies display similar response to rituximab therapy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1094–1097.
- Hospital, M.-A.; Viala, K.; Dragomir, S.; Levy, V.; Cohen-Aubart, F.; Neil, J.; Musset, L.; Choquet, S.; Leger, J.-M.; Leblond, V. Immunotherapy-based regimen in anti-MAG neuropathy: Results in 45 patients. Haematologica 2013, 98, e155–e157.
- Gruson, B.; Ghomari, K.; Beaumont, M.; Garidi, R.; Just, A.; Merle, P.; Merlusca, L.; Marolleau, J.P.; Royer, B. Long-term response to rituximab and fludarabine combination in IgM anti-myelin-associated glycoprotein neuropathy. J. Peripher. Nerv. Syst. 2011, 16, 180–185.
- Massa, F.; Zuppa, A.; Pesce, G.; Demichelis, C.; Bergamaschi, M.; Garnero, M.; Briani, C.; Ferrari, S.; Schenone, A.; Benedetti, L. Bendamustine-rituximab (BR) combined therapy for treatment of immuno-mediated neuropathies associated with hematologic malignancy. J. Neurol. Sci. 2020, 413, 116777.
- Rakocevic, G.; Martinez-Outschoorn, U.; Dalakas, M.C. Obinutuzumab, a potent anti-B-cell agent, for rituximab-unresponsive IgM anti-MAG neuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e460.
- Briani, C.; Visentin, A.; Salvalaggio, A.; Cacciavillani, M.; Trentin, L. Obinutuzumab, a new anti-CD20 antibody, and chlorambucil are active and effective in anti-myelin-associated glycoprotein antibody polyneuropathy. Eur. J. Neurol. 2019, 26, 371–375.
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e720.
- Owen, R.G.; McCarthy, H.; Rule, S.; D’Sa, S.; Thomas, S.K.; Tournilhac, O.; Forconi, F.; Kersten, M.J.; Zinzani, P.L.; Iyengar, S.; et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: A single-arm, multicentre, phase 2 study. Lancet Haematol. 2020, 7, e112–e121.
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050.
- Tam, C.S.; Dimopoulos, M.A.; Garcia-Sanz, R.; Trotman, J.; Opat, S.; Roberts, A.W.; Owen, R.G.; Song, Y.; Xu, W.; Zhu, J.; et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2022, 6, 1296–1308.
- Dimopoulos, M.; Sanz, R.G.; Lee, H.-P.; Trneny, M.; Varettoni, M.; Opat, S.; D’Sa, S.; Owen, R.G.; Cull, G.; Mulligan, S.; et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: A substudy of the phase 3 ASPEN trial. Blood Adv. 2020, 4, 6009–6018.
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations from the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054.
- Castillo, J.J.; Allan, J.N.; Siddiqi, T.; Advani, R.H.; Meid, K.; Leventoff, C.; White, T.P.; Flynn, C.A.; Sarosiek, S.; Branagan, A.R.; et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. J. Clin. Oncol. 2022, 40, 63–71.
- Gertz, M.A.; Dispenzieri, A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA 2020, 324, 79–89.
More
Information
Subjects:
Hematology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
2 times
(View History)
Update Date:
21 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No