Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marion Jost | + 2284 word(s) | 2284 | 2022-03-08 04:31:26 | | | |
| 2 | Conner Chen | -32 word(s) | 2252 | 2022-03-21 04:16:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jost, M. Microbes Living on the Skin. Encyclopedia. Available online: https://encyclopedia.pub/entry/20732 (accessed on 25 June 2026).
Jost M. Microbes Living on the Skin. Encyclopedia. Available at: https://encyclopedia.pub/entry/20732. Accessed June 25, 2026.
Jost, Marion. "Microbes Living on the Skin" Encyclopedia, https://encyclopedia.pub/entry/20732 (accessed June 25, 2026).
Jost, M. (2022, March 18). Microbes Living on the Skin. In Encyclopedia. https://encyclopedia.pub/entry/20732
Jost, Marion. "Microbes Living on the Skin." Encyclopedia. Web. 18 March, 2022.
Copy Citation
The human skin represents the largest human organ. It provides an effective barrier between the human organism and the environment. Superficial skin layers are inhabited by different sorts of microorganisms, such as bacteria, viruses, archaea, and fungi. This heterogeneous community of microorganisms are in mutualistic symbiosis. They play an essential role in the protection against invading pathogens and in the breakdown of natural products. Additionally, they contribute to a special form of innate and adaptive immunity, which links antimicrobial functions and tissue repair.
microbiome
skin
1. Skin Microbiome
The human skin represents the largest human organ. It provides an effective barrier between the human organism and the environment. Superficial skin layers are inhabited by different sorts of microorganisms, such as bacteria, viruses, archaea, and fungi [1]. This heterogeneous community of microorganisms are in mutualistic symbiosis. They play an essential role in the protection against invading pathogens and in the breakdown of natural products. Additionally, they contribute to a special form of innate and adaptive immunity, which links antimicrobial functions and tissue repair [2]. They are able to modulate the production of antimicrobial peptides (AMPs) and various cytokines in and on the skin (e.g., IL-1, IL-17, IL-22, TNF) [1][3]. The genome as a whole, encompassing all of the mentioned microorganisms, is known as the skin microbiome.
Some bacteria are part of the common skin microbiota, e.g., Staphylococcus epidermidis (SE), and ubiquitously colonize human skin, as opposed to others, e.g., Staphylococcus aureus (SA), which are rarely found on healthy human skin [4].
Under normal conditions, transient skin microbes can persist on the skin only for a few hours or days and are not pathogenic (normal immune responses, skin barrier function intact) [1]. If the skin barrier is deficient, or if the microbial balance between commensals and pathogens is disturbed, skin diseases or even systemic diseases can be triggered [2]. Furthermore, it is known that in a chronic inflammatory environment, malignant T-cells are prone to proliferate [5].
2. Cutaneous T-Cell Lymphomas
Primary cutaneous T-cell lymphomas (CTCL) represent a group of non-Hodgkin lymphomas characterized by the accumulation of clonally expanded CD4+ T-cells in the skin. Mycosis fungoides (MF) is the most common subtype and represents around 65–75% of these cases. The etiopathogenesis is still unknown, and a curative treatment option is not yet available. However, disease control can be achieved through stage-adapted therapy, especially in early stage patients [6][7].
CTCL, especially early stages of MF and SS, often resemble chronic inflammatory dermatoses. In particular, the clinical picture is often misinterpreted as eczema or atopic eczema. This often results in a delayed diagnosis for the patient. The incidence of CTCL is currently around 0.4–0.6/100,000/year [7].
The skin lesions in MF are described as erythematous patches and plaques, which can ulcerate, and in advanced stages, patients can develop tumors. SS is an aggressive variant of CTCL that is clinically associated with erythroderma, lymphadenopathy, and leukemia [8][9]. A cure of the disease is currently not possible, but through consistent and stage-adapted therapy, disease control can be achieved. Topical glucocorticosteroids, retinoids, UV therapies, immunosuppressants, radiation therapies, and various immunomodulatory treatments are used for treatment [6][10][11][12][13][14][15][16][17][18][19].
Since the clinical skin changes in CTCL may mimic inflammatory skin disorders, the question was addressed, whether the skin microbiota might play a significant role in the development or in the progression of the disease. In the 1960s and 1970s, the first case reports about the positive effects of antibiotics and antiviral therapy in CTCL were described. Since then, it was suspected that a chronic antigen stimulus might trigger the development and/or the progression of the disease (Figure 1) [20].
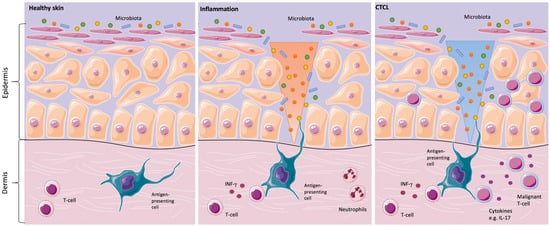
Figure 1. Skins’ interactions with microorganisms in healthy skin, inflammation, and in primary cutaneous T-cell lymphomas (CTCL). Healthy skin in homeostasis (left image). Classical inflammatory responses, as characterized by infiltrating neutrophils and monocytes, alongside interferon-γ (IFN-γ)-producing T-cells. In CTCL, a chronic antigen stimulus from interaction with microorganisms is under suspicion in the origin or/and the progression of the disease (middle image). For instance, Staphylococcal enterotoxin A can stimulate the activation of signal transducer and activator of transcription 3 (STAT3), as shown in in vitro CTCL models, resulting in an upregulation of interleukin IL-17. IL-17 might indirectly influence tumorigenesis by modulating angiogenesis and inflammation (right image) [2][13][21].
In inflammatory skin diseases, both changes in the expression of antimicrobial peptides on the skin surface and changes in microbial colonization are known. Cancer-induced skin barrier defects and potential shifts in the skin microbiome might render CTCL patients prone to increased susceptibility to bacterial infections [16]. This could be of special relevance for CTCL patients since it is common knowledge that SA infections play a major role in morbidity and mortality in these patients [22][23].
3. Microbiome Analysis
The role of the microbiota in CTCL is not well understood yet. The recent establishment of next-generation sequencing methods and reference databases puts the skin microbiome and its role in the pathogenesis of CTCL in the spotlight.
Very recently, two studies on the skin microbiome in CTCL were published. These studies are focused on MF, and a further one on parapsoriasis, respectively. The literature was also checked for data on rarer subtypes of CTCL, e.g., CD4+ small/medium-sized lymphoproliferation and primary cutaneous CD30+ lymphoproliferative disorders. However, there is no evidence published about the microbiome in these entities.
Harkins et al. analyzed skin swabs from four MF patients (stages IA to IIIA) and two SS patients (stage IVA1), matched with samples from age- and sex-matched healthy volunteers. Via “shotgun metagenomic sequencing”, only slight shifts in the skin microbiota were noticed. They observed increasing trends in the mean relative abundances of Corynebacterium species and decreasing trends in Cutibacterium species without statistical significance. The authors suggested that the bacterial shifts may correlate with disease stage or treatment status [21].
Another group, Salava et al., analyzed skin swabs from 20 Patients with MF at stages IA-IIB. They matched their data from both 16S rRNA gene sequencing” and “whole-genome shotgun sequencing” to swabs from contralateral healthy-appearing body sites on the same patients. This group also could not detect significant differences at the genus level or in the microbial diversity in the composition of the skin microbiome in their analyzed patients [24].
In 2017 the same group already looked into microbiome changes in parapsoriasis-affected skin. In comparison to healthy-appearing, contra-lateral skin, there were not any statistically significant differences detectable [25]. Parapsoriasis is primarily considered an inflammatory disease. However, it was discussed that parapsoriasis might represent a precursor to the development of lymphoma. Clinically, patients present persistent, finely scaling, and mildly eczematic lesions that might resemble early stage MF [26].
In summary, the CTCL skin microbiome analyses did not yield statistically significant results, probably due to the small number of patients. CTCL is a rare disease; hence, multicenter analyses, the inclusion of larger patient numbers and investigations according to the same study protocol, should be considered to find statistically significant differences in the future.
3.1. Location Sites
Human skin sites provide diverse microenvironments that vary in pH, temperature, moisture, sebum content, and ultraviolet light exposure. Due to these characteristics, the sites can be grouped in sebaceous (face, chest and back), moist (bend of elbow, back of knee and groin), and dry (volar forearm and palm). Dependent on the physiology of the skin site, the composition of microbial communities changes in the relative abundance of bacterial taxa [2][27][28]. Despite constant environmental change, skin microbial communities are quite stable at least over a two-year time period [29].
3.2. Methods
The skins’ microorganisms are analyzed either by “amplicon sequencing” or “shotgun metagenomics”. In “amplicon sequencing”, the unique “16S ribosomal RNA gene” is sequenced for bacteria, which is called “16S rRNA gene sequencing/analysis”. For fungi, the internal transcribed spacer 1 (ITS1) region of the eukaryotic ribosomal gene is used. This follows assembly or mapping to a reference database. The “16S rRNA gene sequence” is like a unique “barcode” for every microbe. In contrast, “shotgun metagenomics/whole genome sequencing” captures the entire complement of genetic material in a sample without a previous targeted amplification step, either for DNA or RNA, which also includes the hosts’ genetic information [2][27]. An overview of the methods that were applied to CTCL/parapsoriasis samples in recent studies is given in Table 1.
Table 1. Methods and controls used in recent skin microbiome studies.
| Author/Year | Patients | Skin Swabs | 16S rRNA Gene Sequencing | Shotgun Metagenomics | Control Skin Swabs | Statistically Significant Differences |
|---|---|---|---|---|---|---|
| Salava et al./2020 [24] | 20 | lymphoma-affected (MF) |
completed | completed | healthy-appearing, contra-lateral | none detected |
| Salava et al./2017 [25] | 13 | parapsoriasis-affected | completed | not completed | healthy-appearing, contra-lateral | none detected |
| Harkins et al./2020 [21] | 6 | lymphoma-affected (MF/SS) |
not completed | completed | healthy volunteer (lower back, thigh) | none detected |
3.3. Healthy Controls
Matching the results from the microbiome analysis with the contralateral healthy-appearing skin or with the results from a healthy volunteer is essential because the ecological body site is a greater determinant of the microbiota composition than individual genetic variation. This means that the antecubital fossa, back, and plantar heel are more similar to the same site on another individual than to any other site on the same individual [1][27][30]. This knowledge is important for any kind of microbiome analysis and not specific for CTCL patients.
3.4. General Limitations of Skin Microbiome Analyses
It is important to note that there are still limitations in the current microbiome analyses performed by “amplicon sequencing” or “whole-genome metagenomis”. Both cannot differentiate between living and dead microorganisms. To resolve this issue, it might be feasible to pre-digest and remove dead microbial cells from the analysis, to obtain a more accurate assessment of the living microbiome [28].
Skin microbiome analysis usually relies on skin swabs; however, some microorganisms are variably present at the surface compared with deeper skin layers. These issues need to be addressed in future study protocols and are the topic of a recent review by Byrd et al. [2].
4. Bacteria
SA and SE, both members of the Staphylococcus genus, represent the main players when talking about skin microbiota. In CTCL, SA infections are known to play a major role in morbidity and mortality in these patients. However, the question of if the abundance of SA, its toxins, or the cancer-induced skin barrier defects are the major problem is not yet clarified [22][23][31].
4.1. Staphylococcus aureus and Staphylococcus aureus Enterotoxin
SA can generate a pro-oncogenic milieu in lesional skin in vivo. Supporting the hypothesis of a potential relevance of SA, CD4+ T-cell responses to SA can inadvertently enhance neoplastic progression in models of CTCL [22][32].
Staphylococcal enterotoxins (SEs) released by certain lineages of SA act as a class of “superantigens” that are extremely potent activators of T-cells [13][33][34].
The toxins provide access to deeper layers of the skin by disrupting the cell–cell contacts between keratinocytes [4]. The enterotoxins bind directly as whole proteins to major histocompatibility complex class II molecules outside the antigen-peptide binding groove and to certain families of T-cell receptor (TCR) V beta chains crosslinking TCR complexes and inducing T-cell activation at extremely low concentrations [22]. Willerslev-Olsen et al. suggested that toxin-mediated activation of malignant cells do not rely on the expression of a single, toxin-specific TCR-Vb chain in malignant cells but on the expression of multiple toxin binding, TCR-Vb chains expressed in bystander non-malignant tumor-infiltrating T-cells [13].
In CTCL patients, an increased prevalence of human leukocyte antigen (HLA)-DR5 (DR11) and HLA-DQ*03, and in SS HLA-DQ*0502 alleles (odd’s ratio = 7.75) were observed. HLA class II antigens affect the binding of bacterial and present processed antigen to CD4+ T-cells [35]. This may suggest that MF and SS occur in genetically determined hosts [30].
The presence of alpha-toxin further favors the persistence of malignant cells while removing the non-malignant CD4+ T-cells [36]. In 2021, Willerslev-Olsen et al. found evidence that SA and its toxins (SEs) induce the expression of oncogenic miR-155 in CTCL. Furthermore, they were able to show that in two patients with SS, aggressive antibiotic therapy was associated with decreased miR-155 expression in lesional skin [31]. SEs can also activate signal transducer and activator of transcription 3 (STAT3) and induce the expression of IL-17 [13]. Krejsgaard et al. wrote “that it seems plausible that IL-17 indirectly influences CTCL tumorigenesis by modulating angiogenesis and inflammation” [37].
Lindahl et al. summarized in 2021, that via SA toxins triggered effects: “(i) expression of oncogenic miR-155 and regulatory proteins (PD1, FoxP3, and IL-10), (ii) STAT3 and STAT5 activation, (iii) inhibition of anti-tumor cytotoxicity, and (iv) proliferation of primary malignant T-cells in vitro” [33]. According to these results, SA and its toxins might become a therapeutic target in CTCL [36].
4.2. Staphylococcus epidermidis (SE)
Several mechanisms were characterized to describe the effects that SE displays with regard to the skin and the skin barrier.
SE produces lantibiotics (bacteriocins), which are lanthionine containing antibacterial peptides. These induced peptides enhance the skin response via pattern recognition receptors (PRRs), contributing to the initial sensing of microorganisms and intracellular signal transduction [3]. Through the activation of Toll-like receptor 2 (TLR-2), the production of proinflammatory cytokines via TLR-3 is downregulated, and the expression of tight junctions is upregulated [1]. This indicates that innate immune sensing of microbial products does not only involve one TLR ligand but rather a whole pattern of potential ligands [4].
Interestingly, SE can drive skin injury through the secretion of the cysteine protease (EcpA). Its exact role in CTCL needs to be evaluated in the future [20].
4.3. Other Bacteria
The microbiome consists of a large number of different bacteria. In a recent microbiome analysis, Harkins et al. saw increasing trends in the mean relative abundances of Corynebacterium species and decreasing trends in Cutibacterium species. They suggested a possible correlation to the disease stage [21].
Another group reported 10 MF patients with dark brown to black necrotic tumors with eschars [34]. These tumors had Enterococcus cultured from a swab or tissue culture and healed or resolved entirely under appropriate antibiotic therapy [30].
Pseudomonas aeruginosa is considered a factor for a fatal outcome in septic CTCL patients. Together with SA, Pseudomonas aeruginosa is acknowledged to be associated with more than 50% of deaths in this patient group [38]. Whether these bacterial colonizations are primary or secondary findings is not yet clear.
References
- Baviera, G.; Leoni, M.C.; Capra, L.; Cipriani, F.; Longo, G.; Maiello, N.; Ricci, G.; Galli, E. Microbiota in Healthy Skin and in Atopic Eczema. BioMed Res. Int. 2014, 2014, 436921.
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155.
- Naik, S.; Bouladoux, N.; Wilhelm, C.; Molloy, M.J.; Salcedo, R.; Kastenmuller, W.; Deming, C.; Quinones, M.; Koo, L.; Conlan, S.; et al. Compartmentalized control of skin immunity by resident commensals. Science 2012, 337, 1115–1119.
- Bitschar, K.; Wolz, C.; Krismer, B.; Peschel, A.; Schittek, B. Keratinocytes as sensors and central players in the immune defense against Staphylococcus aureus in the skin. J. Dermatol. Sci. 2017, 87, 215–220.
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282.
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714.
- Scarisbrick, J.J.; Quaglino, P.; Prince, H.M.; Papadavid, E.; Hodak, E.; Bagot, M.; Servitje, O.; Berti, E.; Ortiz-Romero, P.; Stadler, R.; et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br. J. Dermatol. 2018, 181, 350–357.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- Scarisbrick, J.J.; Hodak, E.; Bagot, M.; Stranzenbach, R.; Stadler, R.; Ortiz-Romero, P.L.; Papadavid, E.; Evison, F.; Knobler, R.; Quaglino, P.; et al. Blood classification and blood response criteria in mycosis fungoides and Sézary syndrome using flow cytometry: Recommendations from the EORTC cutaneous lymphoma task force. Eur. J. Cancer 2018, 93, 47–56.
- Kempf, W.; Zimmermann, A.K.; Mitteldorf, C. Cutaneous lymphomas-An update 2019. Hematol. Oncol. 2019, 37 (Suppl. S1), 43–47.
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785.
- Harder, J.; Schroder, J.M.; Glaser, R. The skin surface as antimicrobial barrier: Present concepts and future outlooks. Exp. Dermatol. 2013, 22, 1–5.
- Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Litvinov, I.V.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Gniadecki, R.; Mongan, N.P.; Sasseville, D.; et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood 2016, 127, 1287–1296.
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sezary syndrome. Br. J. Dermatol. 2008, 159, 105–112.
- Fanok, M.H.; Sun, A.; Fogli, L.K.; Narendran, V.; Eckstein, M.; Kannan, K.; Dolgalev, I.; Lazaris, C.; Heguy, A.; Laird, M.E.; et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 1116–1125.
- Suga, H.; Sugaya, M.; Miyagaki, T.; Ohmatsu, H.; Kawaguchi, M.; Takahashi, N.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; et al. Skin barrier dysfunction and low antimicrobial peptide expression in cutaneous T-cell lymphoma. Clin. Cancer Res. 2014, 20, 4339–4348.
- Ekman, A.K.; Vegfors, J.; Eding, C.B.; Enerback, C. Overexpression of Psoriasin (S100A7) Contributes to Dysregulated Differentiation in Psoriasis. Acta Derm. Venereol. 2017, 97, 441–448.
- Zasloff, M. Antimicrobial peptides in health and disease. N. Engl. J. Med. 2002, 347, 1199–1200.
- S2k-Guidelines-Cutaneous Lymphomas (ICD10 C82–C86). Available online: https://www.awmf.org/uploads/tx_szleitlinien/032-027l_S2k_Kutane_Lymphome_2021-12.pdf (accessed on 27 December 2021).
- Shelley, W.B. Demethylchlortetracycline and griseofulvin as examples of specific treatment for mycosis fungoides. Br. J. Dermatol. 1981, 104, 477–480.
- Harkins, C.P.; MacGibeny, M.A.; Thompson, K.; Bubic, B.; Huang, X.; Brown, I.; Park, J.; Jo, J.H.; Segre, J.A.; Kong, H.H.; et al. Cutaneous T-Cell Lymphoma Skin Microbiome Is Characterized by Shifts in Certain Commensal Bacteria but not Viruses when Compared with Healthy Controls. J. Investig. Dermatol. 2020, 141, 1604–1608.
- Lindahl, L.M.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Nielsen, P.R.; Blumel, E.; Rittig, A.H.; Celis, P.; Herpers, B.; Becker, J.C.; Stausbol-Gron, B.; et al. Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood 2019, 134, 1072–1083.
- Nguyen, V.; Huggins, R.H.; Lertsburapa, T.; Bauer, K.; Rademaker, A.; Gerami, P.; Guitart, J. Cutaneous T-cell lymphoma and Staphylococcus aureus colonization. J. Am. Acad. Dermatol. 2008, 59, 949–952.
- Salava, A.; Deptula, P.; Lyyski, A.; Laine, P.; Paulin, L.; Väkevä, L.; Ranki, A.; Auvinen, P.; Lauerma, A. Skin Microbiome in Cutaneous T-Cell Lymphoma by 16S and Whole-Genome Shotgun Sequencing. J. Investig. Dermatol. 2020, 140, 2304–2308.e7.
- Salava, A.; Pereira, P.; Aho, V.; Vakeva, L.; Paulin, L.; Auvinen, P.; Ranki, A.; Lauerma, A. Skin Microbiome in Small- and Large-plaque Parapsoriasis. Acta Derm. Venereol. 2017, 97, 685–691.
- Klemke, C.D.; Dippel, E.; Dembinski, A.; Ponitz, N.; Assaf, C.; Hummel, M.; Stein, H.; Goerdt, S. Clonal T cell receptor gamma-chain gene rearrangement by PCR-based GeneScan analysis in the skin and blood of patients with parapsoriasis and early-stage mycosis fungoides. J. Pathol. 2002, 197, 348–354.
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31.
- Amar, Y.; Lagkouvardos, I.; Silva, R.L.; Ishola, O.A.; Foesel, B.U.; Kublik, S.; Schöler, A.; Niedermeier, S.; Bleuel, R.; Zink, A.; et al. Pre-digest of unprotected DNA by Benzonase improves the representation of living skin bacteria and efficiently depletes host DNA. Microbiome 2021, 9, 123.
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64.
- Lewis, D.J. Cutaneous microbiota in the pathogenesis of cutaneous T-cell lymphoma and the role of antibiotic therapy. Int. J. Dermatol. 2020, 59, e223–e224.
- Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Lindahl, L.M.; Buus, T.B.; Pallesen, E.M.H.; Gluud, M.; Bzorek, M.; Nielsen, B.S.; Kamstrup, M.R.; Rittig, A.H.; et al. Staphylococcus aureus Induces Signal Transducer and Activator of Transcription 5—Dependent miR-155 Expression in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2021, 141, 2449–2458.
- Woetmann, A.; Lovato, P.; Eriksen, K.W.; Krejsgaard, T.; Labuda, T.; Zhang, Q.; Mathiesen, A.-M.; Geisler, C.; Svejgaard, A.; Wasik, M.A.; et al. Nonmalignant T cells stimulate growth of T-cell lymphoma cells in the presence of bacterial toxins. Blood 2006, 109, 3325–3332.
- Lindahl, L.M.; Iversen, L.; Ødum, N.; Kilian, M. Staphylococcus aureus and Antibiotics in Cutaneous T-Cell Lymphoma. Dermatology 2021, 1–3.
- Duvic, M.; Feasel, A.M.; Schwartz, C.A.; Cather, J.C. Enterococcal eschars in cutaneous T-cell lymphoma tumors: A distinct clinical entity. Clin. Lymphoma 2000, 1, 141–145.
- Jackow, C.M.; Cather, J.C.; Hearne, V.; Asano, A.T.; Musser, J.M.; Duvic, M. Association of Erythrodermic Cutaneous T-Cell Lymphoma, Superantigen-Positive Staphylococcus aureus, and Oligoclonal T-Cell Receptor Vβ Gene Expansion. Blood 1997, 89, 32–40.
- Blümel, E.; Willerslev-Olsen, A.; Gluud, M.; Lindahl, L.M.; Fredholm, S.; Nastasi, C.; Krejsgaard, T.; Surewaard, B.G.J.; Koralov, S.B.; Hu, T.; et al. Staphylococcal alpha-toxin tilts the balance between malignant and non-malignant CD4+ T cells in cutaneous T-cell lymphoma. Oncoimmunology 2019, 8, e1641387.
- Krejsgaard, T.; Ralfkiaer, U.; Clasen-Linde, E.; Eriksen, K.W.; Kopp, K.L.; Bonefeld, C.M.; Geisler, C.; Dabelsteen, S.; Wasik, M.A.; Ralfkiaer, E.; et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J. Investig. Dermatol. 2011, 131, 1331–1338.
- Lorincz, A.L. Cutaneous T-cell lymphoma (mycosis fungoides). Lancet 1996, 347, 871–876.
More
Information
Subjects:
Oncology; Dermatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
986
Revisions:
2 times
(View History)
Update Date:
21 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No