Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sonia Pernas | + 3034 word(s) | 3034 | 2022-03-10 05:14:06 | | | |
| 2 | Amina Yu | -29 word(s) | 3005 | 2022-03-21 02:42:54 | | | | |
| 3 | Amina Yu | -28 word(s) | 2977 | 2022-03-21 02:47:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pernas, S. Metformin in Breast Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/20722 (accessed on 29 June 2026).
Pernas S. Metformin in Breast Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/20722. Accessed June 29, 2026.
Pernas, Sonia. "Metformin in Breast Cancer" Encyclopedia, https://encyclopedia.pub/entry/20722 (accessed June 29, 2026).
Pernas, S. (2022, March 18). Metformin in Breast Cancer. In Encyclopedia. https://encyclopedia.pub/entry/20722
Pernas, Sonia. "Metformin in Breast Cancer." Encyclopedia. Web. 18 March, 2022.
Copy Citation
Metformin is a widely prescribed oral antidiabetic medication. This biguanide is considered a first-line drug for the management of type 2 diabetes (T2D). Breast cancer is the most prevalent cancer and the leading cause of cancer-related death among women worldwide. Type 2 diabetes–associated metabolic traits such as hyperglycemia, hyperinsulinemia, inflammation, oxidative stress, and obesity are well-known risk factors for breast cancer.
metformin
breast cancer
diabetes
insulin
clinical trials
1. Metformin and the Insulin-Signaling Network
Insulin is a peptide synthesized by pancreatic beta cells in response to increased glucose levels in plasma. Under normal conditions, insulin acts as a regulator of energy storage, metabolism, and growth [1]. IGF1 is an endocrine mediator that regulates cell growth, differentiation, apoptosis, and malignant transformation. The expression of insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1-R) is virtually ubiquitous, thereby explaining the influence of insulin and IGF1 in nearly every tissue. Moreover, both ligands can also bind to alternate receptors with reduced affinity [2][3]. When insulin binds to its receptor, it promotes the phosphorylation of insulin receptor substrate 1 (ISR1) and the subsequent activation of the PI3K/AKT/mTOR signaling cascade. Both hyperglycemia and insulin itself stimulate IGF1 production. Binding of IGF1 to its receptor initiates downstream signaling events that not only involve the PI3K/AKT/mTOR pathway but also the RAS/RAF/MAPK route. These pathways are commonly deregulated in tumor cells, thus providing a mechanistic link between insulin/IGF1 and cancer initiation and progression via enhanced cancer cellular motility and invasion, anaerobic metabolism, dysregulation of epithelial to mesenchymal transition (EMT), tissue inflammation, reactive oxygen species (ROS) production, and angiogenesis, among others [1][4]. Importantly, breast cancer cells express significantly higher IR and IGF1-R levels than normal breast tissues [2][5]. Insulin levels have been related to poor prognosis in breast cancer [6], and persistent hyperinsulinemia reduces levels of IGF Binding Protein 1 (IGFBP-1), thereby increasing the bioactive concentrations of IGF1 [7]. Higher IGF1 levels have been correlated with tumor size and lymph node involvement [5]. Although the role of IGF1-R as a prognostic factor remains controversial, some have linked elevated IGF1-R expression in circulating cancer stem cells (CSCs) to worse prognosis in breast cancer patients [8]. Not surprisingly, pharmacological targeting of the insulin and IGF1 signaling remains an active area of research in oncology.
An ever-growing number of preclinicals have demonstrated that the insulin-sensitizer metformin can block cell proliferation and induce cell cycle arrest and apoptosis in tumor cells [9]. Intriguingly, the potential mechanism(s) of the antitumoral effect of metformin might involve both direct (insulin-independent) and indirect (insulin-dependent) actions [10], which are summarized in Figure 1 and Figure 2. One of the well-accepted insulin-independent effects of metformin involves the activation of the central energy sensor adenosine monophosphate activated protein kinase (AMPK). Activated AMPK downregulates ISR1 and PI3K/AKT/mTOR signaling. Moreover, metformin directly targets respiratory complex I of the electron transport chain in mitochondria, a primary mechanism of action of metformin that reduces energy supply and activates an integrated stress response involving reactive oxygen species (ROS) and DNA damage [11]. Metformin can also induce the expression of REDD1 (Regulated in DNA Damage 1) to inhibit the mTOR pathway via p53, resulting in cyclin D-dependent cell cycle arrest.
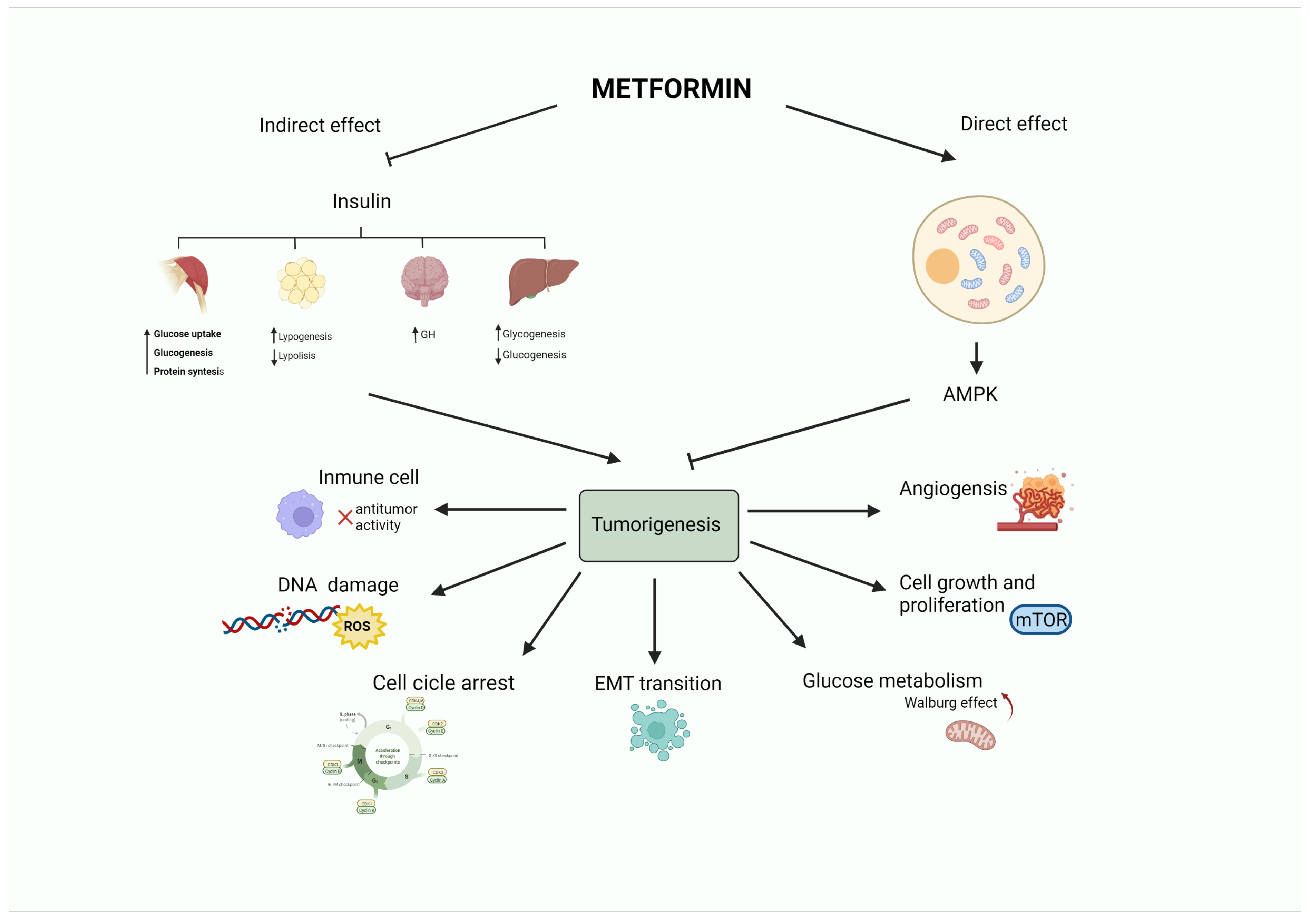
Figure 1. Antitumoral activity of the antidiabetic biguanide metformin. Metformin may affect tumorigenesis by acting on different hallmarks of cancer (angiogenesis, cell growth and proliferation, glucose metabolism, epithelial to mesenchymal transition, cell cycle progression, DNA damage or inflammation). These effects may be led by a direct (insulin-independent) effect mediated by the activation of AMPK. Metformin also has an indirect (insulin-dependent) effect, in which metformin reduces insulin levels. This leads to a decrease in blood glucose by limiting gluconeogenesis and increasing glycogenolysis in the liver, promoting growth hormone synthesis, reducing the release of free fatty acids from adipose tissue, and stimulating lipogenesis, as well as fostering glycogenesis, protein synthesis, and glucose utilization in the muscle.
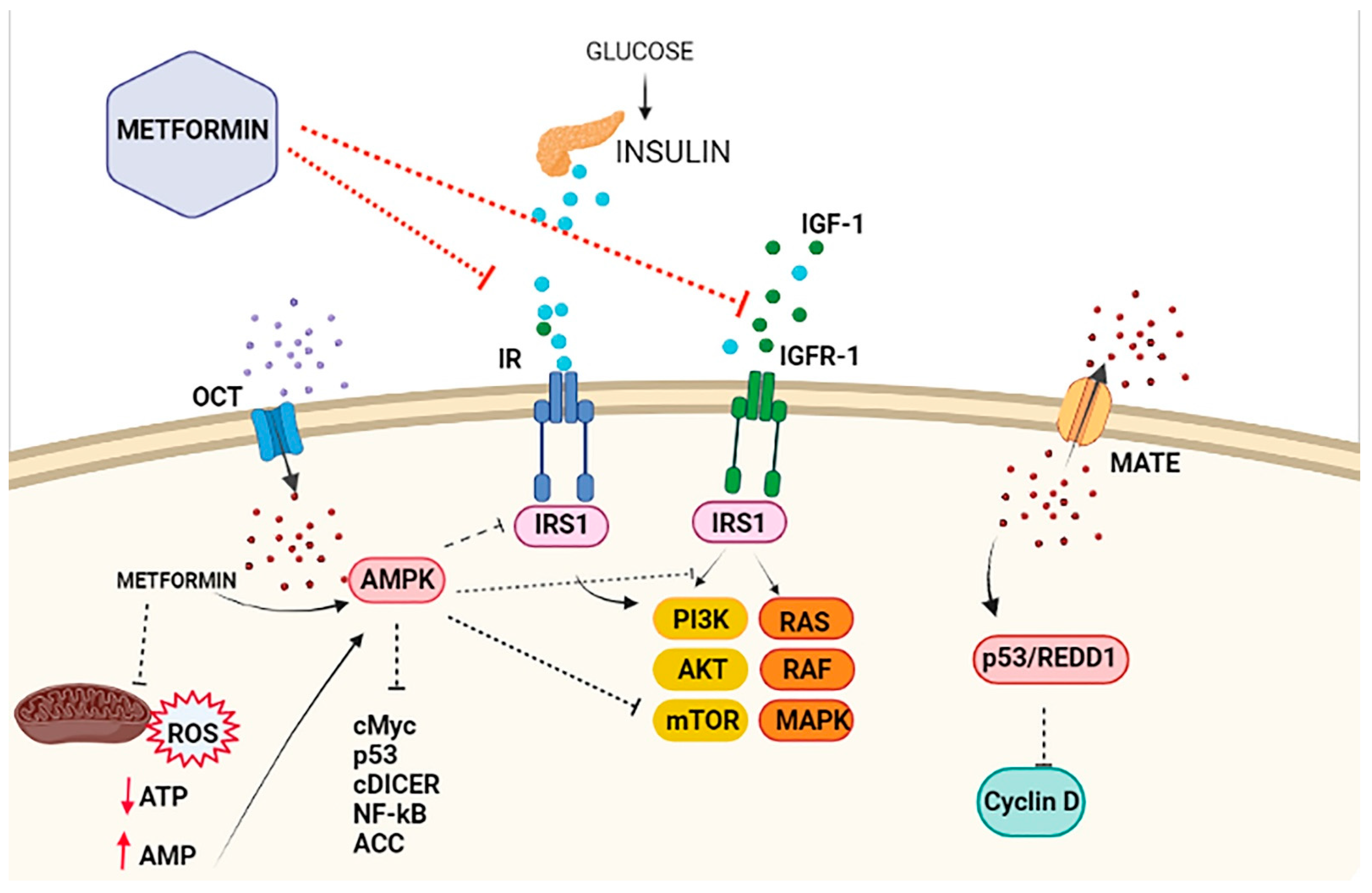
Figure 2. Antiproliferative effects of metformin on breast cancer cells. The antiproliferative activity of metformin in breast cancer is partly attributed to its ability to reduce insulin/IGF1 levels, which inhibits the molecular pathways mediated by them that support tumor initiation and progression (indirect or insulin-dependent mechanism, represented via red lines ˗ ˗ ˗I). Metformin is transported into the cell via the organic cation transporters (OCTs), which support the intracellular accumulation of metformin. On the contrary, the transporters’ multidrug and toxin extrusion (MATE) expel metformin from the cell. Inside the cell, metformin directly activates AMPK and the ‘AMPK dependent’ effects (direct or insulin-independent effects, which are represented via black lines ˗ ˗ ˗I). This process includes the inhibition of IRS1 phosphorylation and blocking of MAPK and mTOR, among other pathways. Metformin is also known to inhibit mitochondrial Complex 1 of the electron transport chain, which reduces ATP levels and increases the AMP/ATP ratio, leading to further AMPK activation.
2. Metformin in Breast Cancer
2.1. Metformin and Prevention of Breast Cancer Incidence and Mortality
Early epidemiologic one evaluating the incidence of cancer in patients on metformin treatment supported a preventative role of metformin in breast cancer [12][13]. However, meta-analyses reached conflicting conclusions [14][15]. Park and colleagues recently reported the results of the Sister, which followed 44,541 women without previous history of breast cancer who had sisters or half-sisters diagnosed with breast cancer. Most of the diabetic population (61%) received metformin. After a median follow-up of 8.6 years, it was failed to identify a relationship between metformin use and overall breast cancer risk (HR 0.98; 95% CI, 0.83–1.15). Metformin use, however, was associated with decreased risk of ER-positive breast cancer (HR 0.86; 95% CI 0.70–1.05), and this inverse association was stronger for a longer duration (≥10 years) of metformin use (HR 0.62; 95% CI, 0.38–1.01; P for trend = 0.09) [16]. These findings suggest an ER status-dependent link between T2D and breast cancer and that the associations between T2D and ER-positive breast cancer might be reduced by long-term usage of metformin [17]. Accordingly, a subpopulation analysis of the Women’s Health Initiative found that metformin use was related to lower risk of ER-positive tumors and fewer human epidermal growth factor receptor 2 (HER2) negative tumors [13]; similarly, from 23 Spanish hospitals observed a protective effect of metformin towards ER-positive/HER2-negative breast cancer [18]. Nevertheless, in others, metformin use was associated with an augmented incidence of ER+ breast carcinomas [19][20], or no differences in breast cancer subtypes attributable to metformin therapy were identified [21][22]. These divergences could be due to differences in metformin exposure and other risk factors such as BMI or age.
It are currently evaluating the preventive effect of metformin in women at high risk of breast cancer. A randomized clinical trial (NCT01793948) is about the effect of metformin in overweight or obese patients at elevated risk of breast cancer based on family history or prior atypical hyperplasia of the breast. Another ongoing trial (NCT01905046) includes patients with a personal history of atypical hyperplasia, carcinoma in situ, breast cancer family history, or high Gail Model Risk. Cells with BRCA gene mutations may be highly dependent on oxidative metabolism, and metformin, via the inhibition of this pathway, may promote a “substrate limitation effect”, interfering with tumor cell proliferation and survival [23]. Because hormonal chemoprevention has been controversial in women with BRCA1 mutations, the ability of metformin to impair both the metabolic rewiring and tumor-initiating capacity of BRCA1 haploinsufficient breast epithelial cells in vitro might suggest new avenues for testing metformin-based chemoprevention strategies in BRCA1 mutation carriers.
Patients with diabetes and breast cancer have up to a 50% higher chance of all-cause mortality than those without diabetes [24][25]. Several have attempted to elucidate the ability of metformin to reduce deaths from breast cancer [26][27][28][29][30]. In an observation involving 919 T2D patients who underwent surgery for breast cancer, the use of metformin attenuated the heightened risk of death observed with prior use of insulin before the diagnosis of breast cancer [31]. Indeed, the majority of the research demonstrates a positive impact of metformin on breast cancer mortality, especially when used in early-stage breast cancer. However, caution is needed, as all these were observational, and their results heterogeneous. Other limitations include no information regarding breast cancer subtypes and tumor stage, inclusion of diabetic populations with different disease severity and duration, and divergent definitions of metformin exposure.
2.2. Metformin and Breast Cancer Treatment
Metformin may enhance the tumoricidal effects of cytotoxic treatments and delay, or even reverse, the occurrence of drug resistance phenomena in breast cancer. In mice models, which have shown a synergistic activity when metformin is combined with paclitaxel, carboplatin, or doxorubicin [32]. For instance, metformin has been found to boost the effect of paclitaxel on AMPK signaling, resulting in a greater downregulation of the mTOR pathway [33]. Alterations in glucose metabolism are characterized by increased glucose uptake, hyperactivated glycolysis, decreased oxidative phosphorylation, and the accumulation of lactate. Targeting key inhibitors of the transporters, enzymes, and mitochondrial components involved in these metabolic alterations may cause tumor sensitizing or resensitizing to chemotherapy. Metformin has been proven to block glucose transporters (GLUT), Lactate dehydrogenase A (LDHA), and complex I of the respiratory chain, and also to decrease citrate in the tricarboxylic acid cycle; this could partly explain improvement in treatment response and countering therapy resistance [34][35].
A combinatorial therapy of low doses of metformin with anthracycline doxorubicin has been shown to kill both tumor-initiating/drug-tolerant CSC and non-CSC in culture and to efficiently prevent tumor relapse in xenograft mouse models [36]. Metformin has also been shown to reduce both tyrosine kinase activity and expression of HER2 in in vitro models of HER2-overexpressing breast cancer cells [37][38][39]. In addition, metformin has been demonstrated to overcome primary resistance to the anti-HER2 monoclonal antibody trastuzumab, both in 3D mammosphere cultures and in HER2-positive breast cancer xenografts [40][41]. This strong experimental basis has provided the rationale for using metformin to improve the treatment of breast cancer patients.
The ability of metformin to directly target mitochondrial respiratory complex I, which reprograms energy production from respiration toward direct glucose utilization, reduces oxygen consumption and increases oxygenation in tumor cells [42]. This primary molecular mechanism of metformin would lead to a glucose concentration–dependent mitigation of the radiation resistance that commonly associates with tumor hypoxia [43][44]. The radiosensitizing mechanisms of metformin may originate not only from re-oxygenation of the hypoxic tumors but also from other cancer cell-intrinsic mechanisms, such as inactivation of mTOR and suppression of the mTOR downstream effectors S6K1 and 4EBP1 [45][46]. Accordingly, a systematic and meta-analysis of retrospective cohort has suggested that metformin might enhance the therapeutic effect of radiotherapy in cancer patients with T2D in terms of short-term efficacy and overall survival (OS) [47]. The ability of metformin to operate as a chemosensitizing/resensitizing agent against various chemotherapeutic drugs, with a differential targeting effect against breast CSC as critical drivers of intrinsic and acquired chemoresistance, it about elsewhere [1][48][49][50][51].
2.2.1. Metformin in Early-Stage Breast Cancer
Several windows of opportunity trials have explored the role of metformin as a single agent in the treatment of early breast cancer. Trials have produced divergent results regarding the ability of two-week neoadjuvant metformin to reduce cellular expression of the proliferative marker Ki-67 in breast cancer tissues [52][53]. Neoadjuvant administration of metformin has been shown to decrease IR expression and suppress PI3K and RAS/MAPK signaling in breast cancer tissues [6][54][55]. It was evaluating the effects of preoperative metformin on immunological factors revealed a significant increasing trend of tumor-infiltrating lymphocytes, CD4+ and CD8+ lymphocytes, and IFNγ in response to metformin, thus suggesting an improved immune function in metformin-treated patients with early breast cancer [56].
It was assessing the impact of neoadjuvant metformin included 2529 patients with early breast cancer treated with primary chemotherapy. The population consisted of 68 diabetic patients on metformin, 87 diabetic patients not taking metformin, and 2374 nondiabetic subjects. Although breast cancer subtypes and tumor stages were well-balanced among the subgroups, a higher postmenopausal status and BMI was observed in the diabetic group. Diabetic patients with breast cancer receiving neoadjuvant metformin and chemotherapy had a higher probability of achieving a pathologic complete response (pCR) than diabetics treated with other antidiabetic drugs (24% vs. 8%) and nondiabetic patients (16%) [57]. This landmark was hypothesis generating and opened the way to prospectively test the potential of metformin as an anti-breast cancer agent.
The METTEN trial (EudraCT number 2011-000490-30) was a randomized multicenter phase II trial to evaluate the efficacy, tolerability, and safety of neoadjuvant chemotherapy and trastuzumab in combination with metformin or placebo for 24 weeks in nondiabetic patients with early-stage HER2-positive breast cancer. The pCR rate was higher in the metformin-containing arm than in the control arm (65.5%, 95% CI: 47.3–80.1 versus 58.6%, 95% CI: 40.7–74.5) but did not reach statistical significance. It lacked power to draw conclusions regarding metformin’s efficacy. However, it is noteworthy that the addition of antidiabetic doses of metformin to neoadjuvant regimens was well tolerated and safe. Thus, only 13% of patients withdrew due to metformin-related gastrointestinal toxicity. The most common grade ≥3 adverse events were neutropenia in both arms and diarrhea in the metformin-containing arm [58]. A analysis of biological correlates in the METTEN, revealed that neoadjuvant metformin significantly decreases the proliferative potential of residual breast cancer cells in those [59] patients that failed to achieve pCR following neoadjuvant therapy. Since the proliferative capacity of residual breast cancer disease informs partial treatment resistance and higher probability of tumor recurrence [60][61], metformin might be considered a safe candidate to target the metabolic peculiarities of residual disease in breast cancer patients.
In the adjuvant scenario, it was exploring the association between metformin use and survival outcomes in diabetic patients with triple-negative breast cancer (TNBC) receiving adjuvant therapy failed to observe any significant impact of the drug. Patients who did not receive metformin and nondiabetic patients tended to have a higher risk of distant metastases compared with the metformin group; however, no significant associations were observed between metformin use and cancer-specific mortality [62]. In HER2-positive breast cancer, a subanalysis of the diabetic population in the ALTTO trial (NCT00490139)—a phase III is about that failed to demonstrate the superiority of lapatinib and trastuzumab compared to trastuzumab as adjuvant treatment—identified a positive effect of metformin in terms of disease-free survival, distant disease-free survival, and OS that was limited to the ER-positive population [63].
The largest prospective trial testing metformin in breast cancer is the recently reported phase III NCIC MA.32 (NCT01101438). This trial investigated the role of metformin in the adjuvant setting in nondiabetic women (N = 3649). The primary endpoint was invasive disease-free survival, and participating subjects were randomly assigned to receive 850 mg of metformin twice daily or placebo for five years. After six months follow-up, two identified metformin-related improvements in weight, BMI, and metabolic variables, as well as significant decreases in circulating plasma estradiol levels [64][65]. Preliminary efficacy results presented at the 2021 San Antonio Breast Cancer Symposium revealed that, irrespective of ER status, the HER2-positive population treated with metformin experienced benefits in terms of invasive disease-free survival (DFS) (HR = 0.64; 95% CI, 0.43–0.95) and OS (HR = 0.53; 95% CI, 0.3–0.98), a benefit not observed in HER2-negative breast cancer patients. Exploratory analyses in HER2-positive subjects suggested that those with at least one “C” allele of the Ataxia Telangiectasia Mutated (ATM) associated Single Nucleotide Polymorphism (SNP) rs11212617 experienced the greatest benefit, which could be considered a possible biomarker of sensitivity to metformin. Regarding toxicity, patients receiving metformin showed a slightly increased incidence of grade ≥3 toxicities, with the most common being gastrointestinal symptoms such as nausea, vomiting, bloating, and diarrhea [66].
2.2.2. Metformin in Metastatic Breast Cancer
The MYME phase II trial evaluated the efficacy of adding metformin to first-line chemotherapy in nondiabetic patients with HER2-negative metastatic breast cancer. Participants were randomized to either liposomal doxorubicin and cyclophosphamide plus metformin 1000 mg twice daily or chemotherapy alone. No benefits in terms of progression-free survival (PFS) or OS were observed in the metformin-containing regimen. However, metformin promoted insulin sensitization and had significant preventive effects on severe, chemotherapy-induced neutropenia [67]. Another phase II trial in which nondiabetic patients with metastatic breast cancer were randomly assigned to receive the investigator’s choice of chemotherapy (anthracycline, platinum, taxane, capecitabine, or vinorelbine) in combination with metformin 850 mg every 12 h or placebo, failed to report significant effects on progression-free survival, response rate, or OS, with a further detrimental impact on quality of life [68].
Despite strong preclinical evidence showing synergistic interactions between metformin and the EGFR tyrosine kinase inhibitor erlotinib in TNBC cell lines [69], a phase I clinical trial of erlotinib and metformin in pretreated metastatic TNBC patients failed to provide significant clinical benefits among the small number of patients included [70]. Overall, and although TNBC is characterized by multiple metabolic aberrations, including PI3K/AKT/mTOR pathway alterations, metformin has not shown any significant clinical benefit in this subset of breast cancer [71][72].
Similarly, regardless of the supporting rationale of targeting mTOR signaling with metformin to circumvent breast cancer endocrine resistance, a randomized phase II (N = 60) of aromatase inhibitors (exemestane or letrozole) plus metformin or placebo in pretreated postmenopausal women with ER+ metastatic breast cancer failed to demonstrate improved efficacy in terms of PFS or OS [73].
Another phase II trial (NCT01627067) evaluated the efficacy and safety of combining metformin with the mTOR inhibitor everolimus and the aromatase inhibitor exemestane in overweight and obese postmenopausal woman with metastatic, ER-positive/HER2-negative breast cancer. This single-arm reported a PFS and OS of 6.3 months (95% CI: 3.8–11.3 months) and 28.8 months (95% CI: 17.5–59.7 months), respectively, similar to those reported in the BOLERO-2 trial (PFS of 6.9 months and OS of 31 months in the everolimus plus exemestane arm). However, one should acknowledge that both involved different population groups in terms of prognosis factors. Thus, the women included in the NCT01627067, it were more heavily pretreated, presenting visceral disease and higher BMI in comparison with the women included in the BOLERO-2 trial [74][75].
The therapeutical potential of PI3K inhibitors targeting the PIK3CA-encoded enzyme p110α, which is known to mediate most if not all cellular responses to insulin, is highly limited due to systemic glucose-insulin compensatory responses capable of reactivating the pathway following inhibition of PI3K activity. Dietary (for example, ketogenic diet) and pharmacological approaches, such as hypoglycemic SGLT2 inhibitors, both capable of suppressing the insulin feedback induced by PI3K inhibitors, have been shown to drastically potentiate the efficacy/toxicity ratios of PI3K inhibitors in animal models [76].
References
- Samuel, S.M.; Varghese, E.; Koklesová, L.; Líšková, A.; Kubatka, P.; Büsselberg, D. Counteracting chemoresistance with metformin in breast cancers: Targeting cancer stem cells. Cancers 2020, 12, 2482.
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191.
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The multifaceted effects of metformin on tumor microenvironment. Semin. Cell Dev. Biol. 2020, 98, 90–97.
- Biello, F.; Platini, F.; D’Avanzo, F.; Cattrini, C.; Mennitto, A.; Genestroni, S.; Martini, V.; Marzullo, P.; Aimaretti, G.; Gennari, A. Insulin/IGF axis in breast cancer: Clinical evidence and translational insights. Biomolecules 2021, 11, 125.
- Dowling, R.J.; Niraula, S.; Chang, M.C.; Done, S.J.; Ennis, M.; McCready, D.R.; Leong, W.L.; Escallon, J.M.; Reedijk, M.; Goodwin, P.J.; et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. 2015, 17, 32.
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419.
- Gennari, A.; Foca, F.; Zamarchi, R.; Rocca, A.; Amadori, D.; De Censi, A.; Bologna, A.; Cavanna, L.; Gianni, L.; Scaltriti, L.; et al. Insulin-like growth factor-1 receptor (IGF-1R) expression on circulating tumor cells (CTCs) and metastatic breast cancer outcome: Results from the TransMYME trial. Breast Cancer Res. Treat. 2020, 181, 61–68.
- Al-Juboori, S.; Vadakekolathu, J.; Idri, S.; Wagner, S.; Zafeiris, D.; Pearson, J.; Almshayakhchi, R.; Caraglia, M.; Desiderio, V.; Miles, A.K.; et al. PYK2 promotes HER2-positive breast cancer invasion. J. Exp. Clin. Cancer Res. 2019, 38, 210.
- Gonzalez-Angulo, A.M.; Meric-Bernstam, F. Metformin: A therapeutic opportunity in breast cancer. Clin. Cancer Res. 2010, 16, 1695–1700.
- Bost, F.; Decoux-Poullot, A.-G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188.
- Col, N.F.; Ochs, L.; Springmann, V.; Aragaki, A.K.; Chlebowski, R.T. Metformin and breast cancer risk: A meta-analysis and critical literature review. Breast Cancer Res. Treat. 2012, 135, 639–646.
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R.; et al. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012, 30, 2844–2852.
- Tang, G.; Satkunam, M.; Pond, G.R.; Steinberg, G.; Blandino, G.; Schünemann, H.J.; Muti, P. Association of metformin with breast cancer incidence and mortality in patients with type II diabetes: A GRADE-assessed systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2018, 27, 627–635.
- Yang, T.; Yang, Y.; Liu, S. Association between metformin therapy and breast cancer incidence and mortality: Evidence from a meta-analysis. J. Breast Cancer 2015, 18, 264–270.
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475.
- Park, Y.-M.M.; Bookwalter, D.B.; O’Brien, K.M.; Jackson, C.L.; Weinberg, C.R.; Sandler, D.P. A prospective study of type 2 diabetes, metformin use, and risk of breast cancer. Ann. Oncol. 2021, 32, 351–359.
- García-Esquinas, E.; Guinó, E.; Castaño-Vinyals, G.; Pérez-Gómez, B.; Llorca, J.; Altzibar, J.M.; Peiró-Pérez, R.; Martín, V.; Moreno-Iribas, C.; Tardón, A.; et al. Association of diabetes and diabetes treatment with incidence of breast cancer. Acta Diabetol. 2016, 53, 99–107.
- Min, W.; Wang, B.; Guo, A.; Mao, G.; Zhao, Y.; Zhang, S.; He, R.; Min, Y.; Huang, Y. The effect of metformin on the clinicopathological features of breast cancer with type 2 diabetes. World J. Oncol. 2020, 11, 23–32.
- Aksoy, S.; Sendur, M.A.N.; Altundag, K. Demographic and clinico-pathological characteristics in patients with invasive breast cancer receiving metformin. Med. Oncol. 2013, 30, 590.
- Besic, N.; Satej, N.; Ratoša, I.; Horvat, A.G.; Marinko, T.; Gazic, B.; Petric, R. Long-term use of metformin and the molecular subtype in invasive breast carcinoma patients—A retrospective study of clinical and tumor characteristics. BMC Cancer 2014, 14, 298.
- Lega, I.C.; Fung, K.; Austin, P.C.; Lipscombe, L.L. Metformin and breast cancer stage at diagnosis: A population-based study. Curr. Oncol. 2017, 24, e85–e91.
- Cuyàs, E.; Fernández-Arroyo, S.; Alarcón, T.; Lupu, R.; Joven, J.; Menendez, J.A. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: Evidence for metformin-based “starvation” strategies in BRCA1 carriers. Oncotarget 2016, 7, 52974–52992.
- Peairs, K.S.; Barone, B.B.; Snyder, C.F.; Yeh, H.-C.; Stein, K.B.; Derr, R.L.; Brancati, F.L.; Wolff, A. Diabetes mellitus and breast cancer outcomes: A systematic review and meta-analysis. J. Clin. Oncol. 2011, 29, 40–46.
- Lawrence, W.R.; Hosler, A.S.; Kuliszewski, M.G.; Leinung, M.C.; Zhang, X.; Schymura, M.J.; Boscoe, F.P. Impact of preexisting type 2 diabetes mellitus and antidiabetic drugs on all-cause and cause-specific mortality among Medicaid-insured women diagnosed with breast cancer. Cancer Epidemiol. 2020, 66, 101710.
- Xu, H.; Chen, K.; Jia, X.; Tian, Y.; Dai, Y.; Li, D.; Xie, J.; Tao, M.; Mao, Y. Metformin use is associated with better survival of breast cancer patients with diabetes: A meta-analysis. Oncologist 2015, 20, 1236–1244.
- He, X.; Esteva, F.J.; Ensor, J.; Hortobagyi, G.N.; Lee, M.-H.; Yeung, S.-C.J. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol. 2012, 23, 1771–1780.
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Blandino, G. Clinical pathological characteristics and prognostic analysis of 1013 breast cancer patients with diabetes. Breast Cancer Res. Treat. 2013, 137, 807–816.
- Oppong, B.A.; Pharmer, L.A.; Oskar, S.; Eaton, A.; Stempel, M.; Patil, S.; King, T.A. The effect of metformin on breast cancer outcomes in patients with type 2 diabetes. Cancer Med. 2014, 3, 1025–1034.
- Zhang, Z.-J.; Yuan, J.; Bi, Y.; Wang, C.; Liu, Y. The effect of metformin on biomarkers and survivals for breast cancer- a systematic review and meta-analysis of randomized clinical trials. Pharmacol. Res. 2019, 141, 551–555.
- Choi, M.; Han, J.; Yang, B.R.; Jang, M.-J.; Kim, M.; Lee, D.-W.; Kim, T.-Y.; Im, S.-A.; Lee, H.-B.; Moon, H.-G.; et al. Association of insulin, metformin, and statin with mortality in breast cancer patients. Cancer Res. Treat. 2021, 53, 65–76.
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201.
- Rocha, G.Z.; Dias, M.M.; Ropelle, E.R.; Osório-Costa, F.; Rossato, F.A.; Vercesi, A.E.; Saad, M.J.; Carvalheira, J.B. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin. Cancer Res. 2011, 17, 3993–4005.
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin targets glucose metabolism in triple negative breast cancer. J. Oncol. Transl. Res. 2018, 4, 129.
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting glucose metabolism to overcome resistance to anticancer chemotherapy in breast cancer. Cancers 2020, 12, 2252.
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511.
- Kim, J.; Lee, J.; Kim, C.; Choi, J.; Kim, A. Anti-cancer effect of metformin by suppressing signaling pathway of HER2 and HER3 in tamoxifen-resistant breast cancer cells. Tumour. Biol. 2016, 37, 5811–5819.
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 2009, 8, 88–96.
- Vázquez-Martín, A.; Oliveras-Ferraros, C.; del Barco, S.; Martín-Castillo, B.; Menéndez, J.A. mTOR inhibitors and the anti-diabetic biguanide metformin: New insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb). Clin. Transl. Oncol. 2009, 11, 455–459.
- Cufí, S.; Corominas-Faja, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Dorca, J.; Barrera, J.B.; Martín-Castillo, B.; Menendez, J.A. Metformin-induced preferential killing of breast cancer initiating CD44+CD24−/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012, 3, 395–398.
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res. Treat. 2011, 126, 355–364.
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res. 2013, 19, 6741–6750.
- Lin, A.; Maity, A. Molecular pathways: A novel approach to targeting hypoxia and improving radiotherapy efficacy via reduction in oxygen demand. Clin. Cancer Res. 2015, 21, 1995–2000.
- Brown, S.L.; Kolozsvary, A.; Isrow, D.M.; Al Feghali, K.; Lapanowski, K.; Jenrow, K.A.; Kim, J.H. A novel mechanism of high dose radiation sensitization by metformin. Front. Oncol. 2019, 9, 247.
- Song, C.W.; Lee, H.; Dings, R.P.M.; Williams, B.; Powers, J.; Dos Santos, T.; Choi, B.-H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362.
- Kim, J.H.; Jenrow, K.A.; Brown, S.L. Novel biological strategies to enhance the radiation therapeutic ratio. Radiat. Oncol. J. 2018, 36, 172–181.
- Rao, M.; Gao, C.; Guo, M.; Law, B.Y.K.; Xu, Y. Effects of metformin treatment on radiotherapy efficacy in patients with cancer and diabetes: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 4881–4890.
- Cioce, M.; Valerio, M.; Casadei, L.; Pulito, C.; Sacconi, A.; Mori, F.; Biagioni, F.; Manetti, C.; Muti, P.; Strano, S.; et al. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget 2014, 5, 4129–4143.
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59.
- Shi, P.; Liu, W.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; Chen, C.; et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017, 3, 17010.
- Qiu, J.; Zheng, Q.; Meng, X. Hyperglycemia and chemoresistance in breast cancer: From cellular mechanisms to treatment response. Front. Oncol. 2021, 11, 628359.
- Bonanni, B.; Puntoni, M.; Cazzaniga, M.; Pruneri, G.; Serrano, D.; Guerrieri-Gonzaga, A.; Trabacca, M.S.; Viale, G.; Bruzzi, P.; DeCensi, A.; et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012, 30, 2593–2600.
- Kalinsky, K.; Zheng, T.; Hibshoosh, H.; Du, X.; Mundi, P.; Yang, J.; Refice, S.; Feldman, S.M.; Taback, B.; Connolly, E.; et al. Proteomic modulation in breast tumors after metformin exposure: Results from a “window of opportunity” trial. Clin. Transl. Oncol. 2017, 19, 180–188.
- Niraula, S.; Dowling, R.; Ennis, M.; Chang, M.C.; Done, S.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830.
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794.
- Tsukioki, T.; Shien, T.; Tanaka, T.; Suzuki, Y.; Kajihara, Y.; Hatono, M.; Kawada, K.; Kochi, M.; Iwamoto, T.; Ikeda, H.; et al. Influences of preoperative metformin on immunological factors in early breast cancer. Cancer Chem. Pharmacol. 2020, 86, 55–63.
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.-C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302.
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; Alvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; Amillano, K.; Domínguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: The METTEN study. Oncotarget 2018, 9, 35687–35704.
- Lopez-Bonet, E.; Buxó, M.; Cuyàs, E.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; et al. Neoadjuvant metformin added to systemic therapy decreases the proliferative capacity of residual breast cancer. J. Clin. Med. 2019, 8, 2180.
- Cabrera-Galeana, P.; Muñoz-Montaño, W.; Lara-Medina, F.; Alvarado-Miranda, A.; Pérez-Sánchez, V.; Villarreal-Garza, C.; Quintero, R.M.; Porras-Reyes, F.; Bargallo-Rocha, E.; Del Carmen, I.; et al. Ki67 changes identify worse outcomes in residual breast cancer tumors after neoadjuvant chemotherapy. Oncologist 2018, 23, 670–678.
- Tokuda, E.; Horimoto, Y.; Arakawa, A.; Himuro, T.; Senuma, K.; Nakai, K.; Saito, M. Differences in Ki67 expressions between pre- and post-neoadjuvant chemotherapy specimens might predict early recurrence of breast cancer. Hum. Pathol. 2017, 63, 40–45.
- Bayraktar, S.; Hernadez-Aya, L.F.; Lei, X.; Meric-Bernstam, F.; Litton, J.K.; Hsu, L.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Effect of Metformin on Survival Outcomes in Diabetic Patientswith Triple Receptor-Negative Breast Cancer. Cancer 2012, 118, 1202–12011.
- Sonnenblick, A.; Agbor-Tarh, D.D.; Bradbury, I.I.; Di Cosimo, S.; Azim, H.A., Jr.; Fumagalli, D.; Sarp, S.S.; Wolff, A.; Andersson, M.M.; Kroep, J.; et al. Impact of diabetes, insulin, and metformin use on the outcome of patients with human epidermal growth factor receptor 2-positive primary breast cancer: Analysis from the ALTTO phase III randomized trial. J. Clin. Oncol. 2017, 35, 1421–1429.
- Goodwin, P.J.; Parulekar, W.R.; Gelmon, K.A.; Shepherd, L.E.; Ligibel, J.A.; Hershman, D.L.; Rastogi, P.; Mayer, I.A.; Hobday, T.J.; Lemieux, J.; et al. Effect of metformin vs. placebo on and metabolic factors in NCIC CTG MA.32. J. Natl. Cancer Inst. 2015, 107, djv006.
- Pimentel, I.; E Chen, B.; Lohmann, A.E.; Ennis, M.; Ligibel, J.; Shepherd, L.; Hershman, D.L.; Whelan, T.; Stambolic, V.; Mayer, I.; et al. The effect of metformin vs. placebo on sex hormones in Canadian cancer trials group MA.32. J. Natl. Cancer Inst. 2021, 113, 192–198.
- Goodwin, P. CCTGMA.32, A Phase III Randomized Double-Blind Placebo Controlled Adjuvant Trial of Metformin (MET) vs. Placebo (PLAC) in Early Breast Cancer (BC): Results of the Primary Efficacy Analysis (Clinical Trials.gov NCT01101438); San Antonio Breast Cancer Symposium: San Antonio, TX, USA, 2021.
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442.
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23.
- Lau, Y.-K.I.; Du, X.; Rayannavar, V.; Hopkins, B.; Shaw, J.; Bessler, E.; Thomas, T.; Pires, M.M.; Keniry, M.; Parsons, R.; et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5, 10503–10517.
- Fenn, K.; Maurer, M.; Lee, S.M.; Crew, K.D.; Trivedi, M.S.; Accordino, M.K.; Hershman, D.L.; Kalinsky, K. Phase 1 study of erlotinib and metformin in metastatic triple-negative breast cancer. Clin. Breast Cancer 2020, 20, 80–86.
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060.
- Sun, X.; Wang, M.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic reprogramming in triple-negative breast cancer. Front. Oncol. 2020, 10, 428.
- Zhao, Y.; Gong, C.; Wang, Z.; Zhang, J.; Wang, L.; Zhang, S.; Cao, J.; Tao, Z.; Li, T.; Wang, B.; et al. A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer. Oncotarget 2017, 8, 84224–84236.
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529.
- Yam, C.; Esteva, F.; Patel, M.M.; Raghavendra, A.S.; Ueno, N.T.; Moulder, S.L.; Hess, K.R.; Shroff, G.S.; Hodge, S.; Koenig, K.H.; et al. Efficacy and safety of the combination of metformin, everolimus and exemestane in overweight and obese postmenopausal patients with metastatic, hormone receptor-positive, HER2-negative breast cancer, a phase II study. Investig. New Drugs 2019, 37, 345–351.
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Publisher Correction: Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
21 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No