 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Brigitte Sola | + 1841 word(s) | 1841 | 2020-09-17 06:23:29 | | | |
| 2 | Bruce Ren | Meta information modification | 1841 | 2020-09-18 03:17:18 | | |
Video Upload Options
Mantle cell lymphoma (MCL) is a rare but aggressive B-cell hemopathy characterized by the translocation t(11;14)(q13;q32) that leads to the overexpression of the cell cycle regulatory protein cyclin D1. This translocation is the initial event of the lymphomagenesis, but tumor cells can acquire additional alterations allowing the progression of the disease with a more aggressive phenotype and a tight dependency on microenvironment signaling. To date, the chemotherapeutic-based standard care is largely inefficient and despite the recent advent of different targeted therapies including proteasome inhibitors, immunomodulatory drugs, tyrosine kinase inhibitors, relapses are frequent and are generally related to a dismal prognosis. As a result, MCL remains an incurable disease.
1. Physiopathology of Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is a rare B-cell lymphoma that represents 5–10% of all non-Hodgkin lymphomas (NHLs), with an incidence of 0.8 cases per 100,000 persons [1]. It develops primarily among elderly individuals with a median age of approximately 67 years and a male-to-female ratio of 2–3:1. At diagnosis, 70% of patients or more have disseminated disease (stage III or IV), with lymphadenopathy (75%), hepato-splenomegaly (35–60%), bone marrow (>60%) and peripheral blood (13–77%) involvement [2]. Waldeyer’s ring and extranodal sites including the gastrointestinal tract, are also frequently involved [3]. The clinical evolution is usually very aggressive, and despite overall response rates above 70% with standard immunochemotherapeutic schemes, few patients can be cured [4].
2. MCL Subtypes
MCL has been recognized as an aggressive small B-cell lymphoma that developed in a linear fashion from naive B-cells. Paradoxically, a subset of patients follows an indolent clinical evolution with a stable disease, and a longer survival, even in the absence of chemotherapy [5], reflecting, in part, that MCL develops along two different pathways. Classical MCL (cMCL) is usually composed of IGHV-unmutated or minimally mutated B-cells that express SOX11 (SRY (sex determining region Y)-box 11), features genetic instability and typically involves lymph nodes and other extranodal sites. Acquisition of additional molecular/cytogenetic abnormalities can lead to even more aggressive, blastoid or pleomorphic MCL. Leukemic non-nodal MCL (nnMCL) develops from IGHV-mutated SOX11−B cells, carrying epigenetic imprints of germinal center (GC)-experienced B cells. It usually involves peripheral blood, bone marrow, and often spleen. These cases feature genetic stability and are frequently clinically indolent; however, secondary abnormalities, often involving TP53, may occur and lead to a very aggressive disease. A third MCL subtype, in situ mantle cell neoplasia (ISMCN), is characterized by the presence of cyclin D1+ cells; most typically in the inner zone of the follicles. Although disseminated, this subtype appears to have a low rate of progression (Figure 1) [3]. Morphologically, three main subtypes of MCL are recognized: the classic, the blastic/blastoid, and the pleomorphic variants. The last two subtypes have higher proliferation rates and are associated with inferior clinical outcome [5].
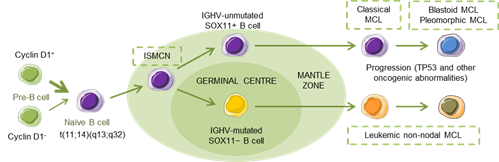
Figure 1. Hypothetical models of major mantle cell lymphoma (MCL) subtypes. Precursor B cells may colonize the inner portion of the mantle zone, representing in situ mantle cell neoplasia (ISMCN). After the introduction of additional genetic and molecular abnormalities, ISMCN may progress, involving or not the transit through the germinal center (GC), to classical MCL or leukemic non-nodal MCL, respectively. More frequently, classical MCL but also leukemic non-nodal MCL undergo additional molecular/cytogenetic abnormalities leading to clinical and sometimes to morphological progression. Adapted from Swerdlow et al.[6].
3. MCL Biological Features and Prognostic Factors
The phenotype of MCL is relatively characteristic with high expression of IgM/IgD surface immunoglobulins. Immunophenotyping reveals that neoplastic cells are usually CD5+ and CD43+ and express the B-cell-associated antigens CD19, CD20, CD22, and CD79. They are usually negative for CD3, CD23, CD11c, CD10, and CD200. MCL cells are generally B-cell lymphoma (BCL)-2 positive and BCL-6 negative [7]. Demonstration of t(11;14)(q13;q32) by FISH or cyclin D1 overexpression by immunohistochemistry is generally required to diagnose MCL, although a small number of cases are cyclin D1−. These cases have a high expression of cyclin D2 or cyclin D3; however, this is not helpful for diagnostics as these proteins are also overexpressed in other B-cell neoplasms. The nuclear SOX11 expression is a highly specific marker for both cyclin D1+/− MCL [8].[8] SOX11 is a transcription factor that has been reported to block terminal B-cell differentiation by regulating PAX5 expression in aggressive MCL. There is also data demonstrating a role for SOX11 as a driver of pro-angiogenic signals in MCL through the regulation of platelet-derived growth factor A, contributing to a more aggressive phenotype [9].
A specific MCL international prognostic index (MIPI) classifies MCL patients into low, intermediate, and high-risk groups, based on four independent prognostic factors: age, Eastern Cooperative Oncology Group (ECOG) performance status, lactate dehydrogenase (LDH), and leukocyte count [10][11]. Other factors such as proliferation of the tumor, karyotypic complexity, genetic aberrations, and DNA methylation are independent prognostic factors for MCL outcome [12].
4. MCL Therapy
Some newly diagnosed MCL patients can be diligently observed, deferring therapy to a later date. Asymptomatic, low tumor burden MCL cases with non-nodal presentation and genetic stability are candidates for this strategy [13]. Delayed treatment in these patients does not adversely affect overall survival (OS) from time of treatment initiation [14]. Although the monoclonal antibody (mAb) anti-CD20 rituximab is considered a standard of care for all newly diagnosed MCL patients, for patients requiring frontline therapy, the initial therapeutic decision is dictated by the age and the fitness of the patient. Since the 1990s, a standard regimen of cyclophosphamide, hydroxydaunomycin (doxorubicin), vincristine, and prednisone (CHOP) has been frequently used to treat MCL patients. Response rates associated with CHOP in this disease are rarely complete or durable, compared with those observed in other B-cell aggressive lymphomas. Therefore, more-intensive strategies have been explored, combining additional agents to improve both the response rates and the durations of response. Induction regimens have included rituximab and high-dose cytarabine (araC) (an antimetabolite pyrimidine analogue), usually followed by autologous stem cell transplantation (ASCT) in younger patients (see below) [15]. The addition of rituximab to CHOP (R-CHOP) was further established as a standard-of-care regimen for the treatment of naive MCL patients. This regimen is now typically administered to patients who are elderly and considered intermediate to high risk, as well as those with relapsed or refractory (R/R) disease, and has been associated with improved OS [16]. However, median survival remains around 5 years, and it is not yet entirely clear how the improved outcomes observed in clinical trial have translated to real-world settings. For patients that achieve remission, consolidation therapy is recommended [17]. For older, less-fit patients there is no generally accepted frontline therapy. R-CHOP regimen followed by rituximab maintenance achieved a significant improvement of OS, with a 4-year survival rate of 87%, largely superior to the 63% survival obtained with interferon (IFN)α therapy [18].
In transplant-ineligible patients with untreated, newly diagnosed MCL, a phase 3 trial demonstrated that frontline bortezomib plus rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP regimen) was associated with a survival benefit over R-CHOP, with a median OS of 90.7 months, significantly longer that the value observed in the R-CHOP group (55.7 months). Therefore, this approach should be considered as a standard of care in this subgroup of patients [19].
Maintenance therapy with rituximab after R-CHOP-based induction has demonstrated clear survival benefit in MCL patients, therefore it represents a well-established approach for postponing disease progression. Among novel agents, the thalidomide-derivative, immunomodulatory drug (IMiD), lenalidomide (Revlimid), has not demonstrated benefit when used as maintenance therapies in MCL, while the first-in-class Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib (Imbruvica®) is still under investigation in these settings [17].
While ASCT is preferentially used in youngest/fit cases as first-line consolidation treatment and almost never employed in the real-cohort patients in R/R MCL [20], allogeneic stem cell transplantation (alloSCT) produces long-term disease-free remissions for around 30–40% patients, especially in younger patients with early relapse or MCL refractory to induction therapy. This approach is considered the sole potentially curative therapy for R/R MCL [21]. In front-line settings, alloSCT was demonstrated to be feasible but should only be considered for patients at high risk of early progression following conventional therapy [22].
Due to the limitations of stem cell transplantation and also considering the relatively poor outcomes associated with chemotherapy, the potential for several chemotherapy-free strategies has been evaluated in MCL patients since early 2000s. Consequently, a growing number of biologically-targeted therapies are profoundly altering the landscape of MCL treatment options in both first-line and relapsed settings [17]. Among these agents, there are currently four drugs licensed across the world: the proteasome inhibitor bortezomib (BTZ, Velcade®), the mechanistic target of rapamycin (mTOR) inhibitor temsirolimus (Torisel®), lenalidomide (Revlimid®), and ibrutinib (Imbruvica®). As single agents, overall response rates (ORRs) are 33% (8% complete response (CR)), 22% (2% CR), 28% (8% CR), and 68% (21% CR), respectively [23][24][25][26]. Beside this clinical efficacy, major differences have been observed in the degree and frequency of adverse events (AEs) associated to these agents in MCL patients. In bortezomib-receiving patients, the most commonly reported AEs are asthenia (72%), peripheral neuropathies (55%), constipation (50%), diarrhea (47%), nausea (44%), and anorexia (39%), the apparition of neuropathy being the most common toxicity, leading to discontinuation and eventually to death [27]. In the case of temsirolimus, hematological toxicity (thrombocytopenia, 72–100%; anemia, 52–66%; neutropenia, 24–77%) is the most frequent AE observed in the clinical setting, and can be generally successfully managed by dose reductions or treatment delay [28]. Hematologic toxicity was also the most common AE observed in R/R MCL patients receiving lenalidomide, with neutropenia and thrombocytopenia observed in 40–62% or 28–12% of the cases, depending on the cohort. Importantly, these effects did not culminate into serious events in any studies, all hematological toxicities being manageable and reversible upon discontinuation of the IMiD [29]. Finally, ibrutinib is by far the safest agent among this list, with hematologic AEs limited to thrombocytopenia (22%), neutropenia (19%), and anemia (18%). Other common AEs including diarrhea (54%), fatigue (50%), nausea (33%), dyspnea (32%), and infection (<10%) were mostly observed during the first 6 months of therapy and with less frequency, thus confirming the safety profile of ibrutinib in R/R MCL [30].
Several novel agents using different target points have also been used with some reported efficacy in R/R MCL. Among the most exciting recent advances in the management of B-cell malignancies, has been the development of chimeric antigen receptor (CAR) T cells [31]. In a recent phase 2 study involving MCL patients who did not respond to BTK inhibitor therapy, the anti-CD19 CAR T-cell therapy, KTE-X19, achieved durable remission in patients with R/R MCL (93% ORR, 67% CR, with progression-free survival (PFS) of 61% and OS of 83% at 1 year) but not without risks: many study participants experienced high-grade cytopenias, infections, and neurologic events [32]. Cyclin-dependent kinase (CDK)4/6 inhibitors (e.g., abemaciclib, palbociclib) are also an attractive therapeutic option given the role of cell-cycle deregulation in the pathogenesis of MCL [33]. Anti-CD20 mAbs, such as ofatumumab [34] and obinutuzumab, [35] have single-agent activity in rituximab-treated patients and are good candidates to be used in combination with other therapies. Moreover, BH3 mimetic-type BCL2 inhibitors such as ABT-199 (venetoclax), phosphatidylinositol 3-kinase (PI3K)δ inhibitors such as idelalisib, histone deacetylase (HDAC) inhibitors (e.g., vorinostat, abexinostat or panobinostat), mTOR inhibitors (e.g., everolimus), or other small molecules including some second-generation BTK inhibitors, are being developed and explored in MCL [36][37][38][39][40]. Finally, the promising activity of anti-CD38 mAb, such as daratumumab in multiple myeloma, has prompted the initiation of studies in other B-cell malignancies, including MCL [41].
References
- Narendranath Epperla; Mehdi Hamadani; Timothy S. Fenske; Luciano J. Costa; Incidence and survival trends in mantle cell lymphoma. British Journal of Haematology 2017, 181, 703-706, 10.1111/bjh.14699.
- Sergio Cortelazzo; Maurilio Ponzoni; Andres Jm Ferreri; Martin Dreyling; Mantle cell lymphoma. Critical Reviews in Oncology/Hematology 2012, 82, 78-101, 10.1016/j.critrevonc.2011.05.001.
- Swerdlow S.H.. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.; World Health Organization. International Agency for Research on Cancer., Eds.; WHO: Geneva, Switzerland, 2017; pp. 1-586.
- Simon Rule; The modern approach to mantle cell lymphoma.. Hematological Oncology 2019, 37, 66-69, 10.1002/hon.2596.
- Pedro Jares; Dolors Colomer; Elías Campo; Molecular pathogenesis of mantle cell lymphoma.. Journal of Clinical Investigation 2012, 122, 3416-23, 10.1172/JCI61272.
- Steven H. Swerdlow; Elías Campo; Stefano A. Pileri; Nancy Lee Harris; Harald Stein; Reiner Siebert; Ranjana Advani; Michele Ghielmini; Gilles Salles; Andrew D. Zelenetz; et al.Elaine S. Jaffe The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375-2390, 10.1182/blood-2016-01-643569.
- Pedro Jares; Elas Campo; Elías Campo; Advances in the understanding of mantle cell lymphoma. British Journal of Haematology 2008, 142, 149-165, 10.1111/j.1365-2141.2008.07124.x.
- Ana Mozos; Cristina Royo; Elena Hartmann; Daphne De Jong; Cristina Baró; Alexandra Valera; Kai Fu; Dennis D. Weisenburger; Jan Delabie; Shih-Sung Chuang; et al.Elaine S. JaffeCarmen Ruiz-MarcellanSandeep DaveLisa RimszaRita BrazielRandy D. GascoyneFrancisco SoléArmando López-GuillermoDolors ColomerLouis M. StaudtAndreas RosenwaldGerman OttPedro JaresElías Campo SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009, 94, 1555-1562, 10.3324/haematol.2009.010264.
- Jara Palomero; Maria Carmela Vegliante; Marta Leonor Rodríguez; Álvaro Eguileor; Giancarlo Castellano; Ester Planas-Rigol; Pedro Jares; Inmaculada Ribera-Cortada; Maria C. Cid; Elías Campo; et al.Virginia Amador SOX11 promotes tumor angiogenesis through transcriptional regulation of PDGFA in mantle cell lymphoma. Blood 2014, 124, 2235-2247, 10.1182/blood-2014-04-569566.
- Eva Hoster; Martin Dreyling; Wolfram Klapper; Christian Gisselbrecht; Achiel Van Hoof; Hanneke C. Kluin-Nelemans; Michael Pfreundschuh; Marcel Reiser; Bernd Metzner; Hermann Einsele; et al.Norma PeterWolfram JungBernhard WörmannWolf-Dieter LudwigUlrich DührsenHartmut EimermacherHannes WandtJoerg HasfordWolfgang HiddemannMichael Unterhaltfor the German Low Grade Lymphoma Study Group (GLSG) and the European Mantle Cell Lymphoma Network A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558-565, 10.1182/blood-2007-06-095331.
- Julie M. Vose; Mantle cell lymphoma: 2015 update on diagnosis, risk-stratification, and clinical management. American Journal of Hematology 2015, 90, 739-745, 10.1002/ajh.24094.
- Ana C. Queirós; Renée Beekman; Roser Vilarrasa-Blasi; Martí Duran-Ferrer; Guillem Clot; Angelika Merkel; Emanuele Raineri; Nuria Russiñol; Giancarlo Castellano; Sílvia Beà; et al.Alba NavarroMarta KulisNúria Verdaguer-DotPedro JaresAnna EnjuanesMaría José CalasanzAnke BergmannInga VaterItziar SalaverríaHarmen Van De WerkenWyndham H. WilsonAvik DattaPaul FlicekRomina RoyoJoost MartensEva GinéArmando Lopez-GuillermoHendrik G. StunnenbergWolfram KlapperChristiane PottSimon HeathIvo G. GutReiner SiebertElías CampoJosé I. Martín-SuberoNuria Russiño Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 2016, 30, 806-821, 10.1016/j.ccell.2016.09.014.
- Verònica Fernàndez; Olga Salamero; B. Espinet; Francesc Solé; Cristina Royo; Alba Navarro; Francisca Camacho; Sílvia Beà; Elena Hartmann; Virginia Amador; et al.Luis HernándezClaudio AgostinelliRachel L SargentMaria RozmanMarta AymerichDolors ColomerNeus VillamorSteven H. SwerdlowStefano A PileriF. Xavier BoschMiguel A. PirisEmili MontserratGerman OttA RosenwaldArmando López-GuillermoPedro JaresSergi SerranoElías Campo Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma.. Cancer Research 2010, 70, 1408-18, 10.1158/0008-5472.CAN-09-3419.
- Peter Martin; Amy Chadburn; Paul Christos; Karen Weil; Richard R. Furman; Jia Ruan; Rebecca Elstrom; Ruben Niesvizky; Scott Ely; Maurizio DiLiberto; et al.Ari MelnickDaniel M. KnowlesSelina Chen-KiangMorton ColemanJohn P. Leonard Outcome of Deferred Initial Therapy in Mantle-Cell Lymphoma. Journal of Clinical Oncology 2009, 27, 1209-1213, 10.1200/jco.2008.19.6121.
- Stephen E. Spurgeon; Brian G. Till; Peter Martin; Andre H. Goy; Martin P. Dreyling; Ajay K. Gopal; Michael Leblanc; John P. Leonard; Jonathan W. Friedberg; Lawrence Baizer; et al.Richard F. LittleBrad S. KahlMitchell R. Smith Recommendations for Clinical Trial Development in Mantle Cell Lymphoma. JNCI: Journal of the National Cancer Institute 2016, 109, djw263, 10.1093/jnci/djw263.
- Benjamin T. Diamond; Anita Kumar; Mantle Cell Lymphoma. Hematology/Oncology Clinics of North America 2019, 33, 613-626, 10.1016/j.hoc.2019.03.002.
- Fengting Yan; Ajay K. Gopal; Solomon A. Graf; Targeted Drugs as Maintenance Therapy after Autologous Stem Cell Transplantation in Patients with Mantle Cell Lymphoma. Pharmaceuticals 2017, 10, 28, 10.3390/ph10010028.
- Hanneke C. Kluin-Nelemans; Eva Hoster; Olivier Hermine; Jan Walewski; Marek Trneny; C.H. Geisler; S. Stilgenbauer; C. Thiéblemont; U. Vehling-Kaiser; J.K. Doorduijn; et al.B. CoiffierR ForstpointnerH. TillyL. KanzP. FeugierM. SzymczykM. HallekS. KremersG. LepeuL. SanhesJ.M. ZijlstraR. BouabdallahP.J. LugtenburgM. MacroMichael PfreundschuhVít ProcházkaFrancesco Di RaimondoVincent RibragM. UppenkampM. AndréWolfram KlapperW. HiddemannM. UnterhaltM.H. Dreyling Treatment of Older Patients with Mantle-Cell Lymphoma. New England Journal of Medicine 2012, 367, 520-531, 10.1056/nejmoa1200920.
- Tadeusz Robak; Jie Jin; Halyna Pylypenko; Gregor Verhoef; Noppadol Siritanaratkul; Johannes Drach; Markus Raderer; Jiri Mayer; Juliana Pereira; Gayane Tumyan; et al.Rumiko OkamotoSusumu NakaharaPeter HuCarlos AppianiSepideh NematFranco CavalliAchiel Van HoofAdriana SheligaAdriana TeixeiraAkihiro TomitaAlbert Oriol RocafigueraAlexander SuvorovAlexy KuzminAli KhojastehAmel MezliniAnatoly GolenkovAndre BoslyAndrew BelchAnn Van De VeldeÁrpád IllesAshis MukhopadhyayBalkis MeddebBernard De PrijckBernardo GarichocheaBulent UndarCaballero GabarrónCarmen CaoCarmino SouzaCharles FarberCheol Won SuhCristina Ileana BurcoveanuCristina Ligia CebotaruCristina-Ligia TruicaDai MaruyamaDavid BeladaDina Ben YehudaDmitry UdovitsaDoloresEnrica MorraErnst Späth-SchwalbeEva Gonzalez BarcaEvgenii OsmanovFrancisco Javier CapoteFritz OffnerGalvez CardenasGeorg HeßGeorgii ManikhasGovind BabuGrigoriy RekhtmanGuiseppe RossiHerlander MarquesHoria BumbeaHuaqing WangHuiqiang HuangIlseung ChoiIrina BulavinaIrina LysenkoIrit AviviIryna KryachokJan Maciej ZauchaJan NovakJoaquín DíazJudit DemeterJulia AlexeevaJun ZhuKateryna VilchevskayaKenichi IshizawaKenny MauricioKensei TobinaiKiyoshi AndoKudrat AbdulkadryrovLee-Yung ShihLyudmila KuzinaMahmut GumusMaike De WitMarcelo CapraMargarida MarquesMarina GolubevaM. Ojeda-UribeMaryna KyselyovaMasafumi TaniwakiMassimo FedericoMichael CrumpMichele BaccaraniMichinori OguraMiklós EgyedMiklos UdvardyMitsutoshi KurosawaNaokuni UikeNuriet KhuagevaOfer ShpilbergOleg GladkovOlga SamoilovaOlga SerdukPatricia SantiPierre ZacheePolina KaplanRazvan StoiaRémy GressinReyes ArranzRichard GreilSebastian GrosickiSergio CanceladoSreejith NairSteven Le GouillSteven Van SteenweghenSung-Soo YoonSuporn ChuncharuneTatiana ScheiderTatsu ShimoyamaTing LiuTomohiro KinoshitaToshiki UchidaUdomsak BunworasateUmberto VitoloViacheslav PavlovVijay Rao PhooshkooruVladimir LimaVladimir MerkulovWeerasak NawarawongXiaonan HongXiaoyan KeYasuhito TeruiYeow Tee GohYoshiharu MaedaYuankai ShiYuri DunaevYurii LorieZhao WangZhixiang ShenZita BorbenyiZoltán GasztonyiZvenyslava MasliakLYM-3002 investigators Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. The Lancet Oncology 2018, 19, 1449-1458, 10.1016/s1470-2045(18)30685-5.
- Erden Atilla; Pinar Ataca Atilla; Taner Demirer; Current treatment strategies in relapsed/refractory mantle cell lymphoma: where are we now?. International Journal of Hematology 2016, 105, 257-264, 10.1007/s12185-016-2164-2.
- Jennifer E. Vaughn; Mohamed L. Sorror; Barry E. Storer; Thomas R. Chauncey; Michael A. Pulsipher; Richard T. Maziarz; Michael B. Maris; Parameswaran Hari; Ginna G. Laport; Georg N. Franke; et al.Edward D. AguraAmelia A. LangstonAndrew R. RezvaniRainer StorbBrenda M. SandmaierDavid G. Maloney Long-term sustained disease control in patients with mantle cell lymphoma with or without active disease after treatment with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning.. Cancer 2015, 121, 3709-16, 10.1002/cncr.29498.
- Simon Rule; Gordon Cook; Nigel H. Russell; Ann Hunter; Stephen Robinson; Nick Morley; Anna Sureda; Pip Patrick; Laura Clifton-Hadley; Toyin Adedayo; et al.Amy KirkwoodKarl S. Peggs Allogeneic stem cell transplantation as part of front line therapy for Mantle cell lymphoma. British Journal of Haematology 2018, 184, 999-1005, 10.1111/bjh.15723.
- Richard I. Fisher; Steven H. Bernstein; Brad S. Kahl; Benjamin Djulbegovic; Michael J. Robertson; Sven De Vos; Elliot Epner; Amrita Krishnan; John P. Leonard; Sagar Lonial; et al.Edward A. StadtmauerOwen A. O'connorHongliang ShiAnthony L. BoralAndré Goy Multicenter Phase II Study of Bortezomib in Patients With Relapsed or Refractory Mantle Cell Lymphoma. Journal of Clinical Oncology 2006, 24, 4867-4874, 10.1200/jco.2006.07.9665.
- Georg Hess; Raoul Herbrecht; Jorge Romaguera; Gregor Verhoef; Michael Crump; Christian Gisselbrecht; Anna Laurell; Fritz Offner; Andrew Strahs; Anna Berkenblit; et al.Orysia HanushevskyJill ClancyBecker HewesLaurence MooreBertrand Coiffier Phase III Study to Evaluate Temsirolimus Compared With Investigator's Choice Therapy for the Treatment of Relapsed or Refractory Mantle Cell Lymphoma. Journal of Clinical Oncology 2009, 27, 3822-3829, 10.1200/jco.2008.20.7977.
- Andre Goy; Rajni Sinha; Michael E. Williams; Sevgi Kalayoglu Besisik; Johannes Drach; Radhakrishnan Ramchandren; Lei Zhang; Sherri Cicero; Tommy Fu; Thomas E. Witzig; et al. Single-Agent Lenalidomide in Patients With Mantle-Cell Lymphoma Who Relapsed or Progressed After or Were Refractory to Bortezomib: Phase II MCL-001 (EMERGE) Study. Journal of Clinical Oncology 2013, 31, 3688-3695, 10.1200/jco.2013.49.2835.
- Michael Wang; Simon Rule; Peter Martin; Andre Goy; Rebecca Auer; Brad S. Kahl; Wojciech Jurczak; Ranjana H. Advani; Jorge E. Romaguera; Michael E. Williams; et al.Jacqueline BarrientosEwa ChmielowskaJohn RadfordStephan StilgenbauerMartin DreylingWieslaw Wiktor JędrzejczakP. W. M. JohnsonStephen E. SpurgeonLei LiLiang ZhangKate NewberryZhishuo OuNancy ChengBingliang FangJesse McGreivyFong ClowJoseph J. BuggyBetty Y. ChangDarrin M. BeaupreLori A. KunkelKristie A. Blum Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma.. New England Journal of Medicine 2013, 369, 507-16, 10.1056/NEJMoa1306220.
- Robert C. Kane; Ramzi Dagher; Ann Farrell; Chia-Wen Ko; Rajeshwari Sridhara; Robert Justice; Richard Pazdur; Bortezomib for the Treatment of Mantle Cell Lymphoma. Clinical Cancer Research 2007, 13, 5291-5294, 10.1158/1078-0432.ccr-07-0871.
- Krimo Bouabdallah; Vincent Ribrag; Louis Terriou; Jean-Charles Soria; Richard Delarue; Temsirolimus in the treatment of mantle cell lymphoma. Current Opinion in Oncology 2013, 25, S1-S12, 10.1097/cco.0b013e32835de8ee.
- Madhav Desai; Kate Newberry; Zhishuo Ou; Michael Wang; Liang Zhang; Lenalidomide in relapsed or refractory mantle cell lymphoma: overview and perspective. Therapeutic Advances in Hematology 2014, 5, 91-101, 10.1177/2040620714532124.
- Michael L. Wang; Kristie A. Blum; Peter Martin; Andre Goy; Rebecca Auer; Brad S. Kahl; Wojciech Jurczak; Ranjana H. Advani; Jorge E. Romaguera; Michael E. Williams; et al.Jacqueline C. BarrientosEwa ChmielowskaJohn RadfordStephan StilgenbauerMartin DreylingWieslaw Wiktor JedrzejczakP. W. M. JohnsonStephen E. SpurgeonLiang ZhangLinda BaherMei ChengDana LeeDarrin M. BeaupreSimon Rule Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood 2015, 126, 739-745, 10.1182/blood-2015-03-635326.
- James N. Kochenderfer; Steven A. Feldman; Yangbing Zhao; Hui Xu; Mary A. Black; Richard A. Morgan; Wyndham H. Wilson; Steven A. Rosenberg; Construction and Preclinical Evaluation of an Anti-CD19 Chimeric Antigen Receptor. Journal of Immunotherapy 2009, 32, 689-702, 10.1097/cji.0b013e3181ac6138.
- Michael Wang; Javier Munoz; Andre Goy; Frederick L. Locke; Caron A. Jacobson; Brian T. Hill; John M. Timmerman; Houston Holmes; Samantha Jaglowski; Ian W. Flinn; et al.Peter A. McSweeneyDavid B. MiklosJohn M. PagelMarie-Jose KerstenNoel MilpiedHenry FungMax S. ToppRoch HouotAmer BeitinjanehWeimin PengLianqing ZhengJohn M. RossiRajul K. JainArati V. RaoPatrick M. Reagan KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma.. New England Journal of Medicine 2020, 382, 1331-1342, 10.1056/NEJMoa1914347.
- John P. Leonard; Ann S. LaCasce; Mitchell R. Smith; Ariela Noy; Lucian R. Chirieac; Scott J. Rodig; Jian Q. Yu; Shankar Vallabhajosula; Heiko Schoder; Patricia English; et al.Nna S. NeubergPeter MartinMichael M. MillensonScott A. ElyRachel CourtneyNaveed ShaikKeith D. WilnerSophia RandolphAnnick D. Van Den AbbeeleSelina Y. Chen-KiangJeffrey T. YapGeoffrey I. Shapiro Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012, 119, 4597-4607, 10.1182/blood-2011-10-388298.
- Michelle Furtado; Martin J. S. Dyer; Rod Johnson; Margie Berrow; Simon Rule; Ofatumumab monotherapy in relapsed/refractory mantle cell lymphoma - a phase II trial. British Journal of Haematology 2014, 165, 575-578, 10.1111/bjh.12769.
- Franck Morschhauser; Guillaume Cartron; Catherine Thieblemont; Philippe Solal-Céligny; C. Haioun; Reda Bouabdallah; Pierre Feugier; Krimo Bouabdallah; Elina Asikanius; GuiYuan Lei; et al.Michael WengerElisabeth Wassner-FritschGilles Salles Obinutuzumab (GA101) Monotherapy in Relapsed/Refractory Diffuse Large B-Cell Lymphoma or Mantle-Cell Lymphoma: Results From the Phase II GAUGUIN Study. Journal of Clinical Oncology 2013, 31, 2912-2919, 10.1200/jco.2012.46.9585.
- Matthew S. Davids; Andrew W. Roberts; John F. Seymour; John M. Pagel; Brad S. Kahl; William G. Wierda; Soham Puvvada; Thomas J. Kipps; Mary Ann Anderson; Ahmed Hamed Salem; et al.Martin DunbarMing ZhuFranklin PealeJeremy A. RossLori GressickMonali DesaiSu Young KimMaria VerdugoRod A. HumerickhouseGary B. GordonJohn F. Gerecitano Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. Journal of Clinical Oncology 2017, 35, 826-833, 10.1200/jco.2016.70.4320.
- Brad S. Kahl; Stephen E. Spurgeon; Richard R. Furman; I. W. Flinn; Steven E. Coutre; Jennifer R. Brown; N M. Benson; John C. Byrd; Sissy Peterman; Yoonjin Cho; et al.Albert YuWayne R. GodfreyNina D. Wagner-Johnston A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398-3405, 10.1182/blood-2013-11-537555.
- Ulrike Heider; Martin Kaiser; Jan Sterz; Ivana Zavrski; Christian Jakob; Claudia Fleissner; Jan Eucker; Kurt Possinger; Orhan Sezer; Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphoma. European Journal of Haematology 2006, 76, 42-50, 10.1111/j.1600-0609.2005.00546.x.
- Andrew M. Evens; Sriram Balasubramanian; Julie M. Vose; Wael Harb; Leo I. Gordon; Robert Langdon; Julian Sprague; Mint Sirisawad; Chitra Mani; Jeanne Yue; et al.Ying LuanSharon HortonThorsten GraefNancy L. Bartlett A Phase I/II Multicenter, Open-Label Study of the Oral Histone Deacetylase Inhibitor Abexinostat in Relapsed/Refractory Lymphoma. Clinical Cancer Research 2015, 22, 1059-1066, 10.1158/1078-0432.ccr-15-0624.
- Christoph Renner; Pier Luigi Zinzani; Rémy Gressin; Dirk Klingbiel; Pierre-Yves Dietrich; Felicitas Hitz; Mario Bargetzi; Walter Mingrone; Giovanni Martinelli; Andreas Trojan; et al.Krimo BouabdallahAndreas LohriEmmanuel GyanChristine BiaggiSergio CogliattiFrancesco BertoniMichele GhielminiPeter BrauchliNicolas Ketterer A multicenter phase II trial (SAKK 36/06) of single-agent everolimus (RAD001) in patients with relapsed or refractory mantle cell lymphoma. Haematologica 2012, 97, 1085-1091, 10.3324/haematol.2011.053173.
- Chan Y. Cheah; John F. Seymour; Michael L. Wang; Mantle Cell Lymphoma. Journal of Clinical Oncology 2016, 34, 1256-1269, 10.1200/jco.2015.63.5904.

