 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia V. Belosludtseva | + 8167 word(s) | 8167 | 2020-09-16 07:46:28 | | | |
| 2 | Camila Xu | -1 word(s) | 8166 | 2020-10-26 10:13:32 | | |
Video Upload Options
Diabetes mellitus is a chronic disease that is characterized by an absolute or relative deficiency of insulin, the hormone that stimulates the transport of glucose across cell membranes, which leads to an increase in blood glucose—hyperglycemia. Two main types of diabetes are distinguished. Type I diabetes mellitus (about 10% of cases of diabetes) is an autoimmune disorder that results from the progressive destruction of the insulin-producing beta cells of the pancreas by T cells and activated macrophages and eventually leads to insulin deficiency in the organism. It is well known that type I diabetes most frequently develops in childhood and causes severe long-term complications, including retinopathy, neuropathy, and nephropathy [4,5,6]. Type II diabetes or adult-onset diabetes (about 90% of cases) is characterized by an impairment of homeostasis of glucose and insulin, in particular, the development of insulin resistance of target tissues associated with compensatory hyperinsulinemia, followed by beta-cell dysfunction. Type II diabetes mellitus is accompanied by glucose toxicity, lipotoxicity, and chronic oxidative stress, which finally can result in damage to vital organs and development of life-threatening secondary complications [4,7]. Mitochondria are one of the main targets of diabetes at the intracellular level. Recent data indicate that disturbances in mitochondrial calcium transport systems and a pathophysiological phenomenon called the permeability transition pore are involved in the pathogenesis of diabetes mellitus.
1. Mechanisms of the Diabetes-Induced Mitochondrial Dysfunction
Mitochondrial dysfunction refers to alterations in the ultrastructure and functioning of mitochondria, which can occur due to a disturbance of synthetic processes in the cell or the direct action of the damaging agents on the organelles. The crucial characteristics of mitochondrial dysfunction are decreased numbers of mitochondria in tissues, profound ultrastructural abnormalities of the organelles, impaired mitochondrial biogenesis, reduced activity of mitochondrial multienzyme complexes, and suppressed ATP synthesis. Along with other features, mitochondrial dysfunction is also characterized by the disturbance of calcium homeostasis, excessive ROS production, and the MPT pore opening, which can trigger irreversible damage to cell structures and cause apoptotic cell death. Importantly, all these processes take place in the course of type 1 and 2 diabetes mellitus, indicating mitochondrial dysfunction being implicated in the pathogenesis of the disease. The diabetes-related mitochondrial alterations in different tissues and organs of animals and humans are summarized in Table 1.
Table 1. The mechanisms of diabetes-induced mitochondrial dysfunction in vital organs and tissues of human and animals.
| Pancreatic Islets | Heart | Skeletal Muscles | Brain | Kidney | Adipose | Liver | |
|---|---|---|---|---|---|---|---|
| Biogenesis | ↓ [11] ↑ [12] |
↓↑ [13] | ↓ [14] | ↓ [15] | ↓ [16] | ↓ [17] | ↓ [18] ↑ [19] |
| Mitophagy | ↓ [20] ↑ [21] |
↓ [22] | ↓ [23] | ↓ [24] | ↓ [25] ↑ [26] |
↑ [27] | ↓ [28] |
| Fission/Fusion | ↑/↓ [29] | ↑/↓ [30] | ↑ [31] ↓ [32] |
↑/↓ [33] | ↑/↓ [34] | ↑ [35] ↓? |
↑/↓ [36] |
| OXPHOS | ↑ or ↓ or not changed, see review [37] | ||||||
| ROS production | ↑ [20] | ↑ [38] | ↑ [39] | ↑ [40] | ↑ [34] | ↑ [17] | ↑ [41] |
As one can see from Table 1, the rate of mitochondrial ROS production increases in most vital organs and tissues of diabetic animals and patients, while changes in mitochondrial biogenesis, dynamics, mitophagy, and oxidative phosphorylation in diabetes can be tissue-specific. Depending on the animal model used, diabetes-induced changes in some mitochondrial processes in the same tissues can be oppositely directed. Indeed, numerous animal models of diabetes are available, including genetic or spontaneously induced and experimentally induced models. Models of spontaneous type I diabetes target single or multiple genes and include a number of stains and sub-stains of rodents with genetic susceptibility for the pathology, including nonobese diabetic (NOD) mice, diabetic Bio-Breeding (BB) rats, Akita mice, Komeda Diabetes Prone (KDP) rats, LETL/KDP rats and substrains, LEW.1AR1/Ztm-iddm rats, etc. The chemical agents streptozotocin and alloxan are also commonly used to induce type I diabetes in rodents, but the regimes for their administration to animals differ significantly. Type 2 diabetes, typically accompanied by obesity, is induced by feeding a high-fat diet to animals, while diabetogenic drugs (streptozotocin, etc.) are often additionally used (for more details see [42,43,44]). Type 2 diabetes can also be produced in rodents by utilizing monogenic mutations (Lep ob/ob (ob/ob) mice, Lepr db/db (db/db) mice, Zucker diabetic fatty (ZDF-Lepr fa/fa or fa/fa) rats), or polygenic mutations (Kuo Kondo (KK) mice, Otsuka Long-Evans Tokushima Fat (OLEFT) rats, etc.). Models of spontaneous type II diabetes are wildly used both in a combination with obesity (Lep ob/ob (ob/ob) mice, etc.) and in the absence of obesity (Goto-Kakizaki (GK) rats, etc.). On the one hand, the pathophysiological and genetic features of animal models can affect the above processes in mitochondria. On the other hand, it is equally important to pay attention to the stage of development of diabetes mellitus (or the duration of its induction). For example, in humans, mitophagy in pancreatic β-cells is activated at the pre-diabetic stage, but significantly suppressed during diabetes [20]. In animal models, responses to exposure to diabetogenic drugs and high-energy diet might be the result of both adaptive and maladaptive processes, which, unfortunately, are often difficult to separate.
1.1. Imbalance of Mitochondrial Biogenesis and Mitophagy upon Diabetes Mellitus
The maintenance of the content and structure of mitochondria in the cell (mitochondrial homeostasis) is ensured by the coordination of two opposite processes—mitochondrial biogenesis and mitophagy.
Mitochondrial biogenesis is a complex and highly regulated process that requires the coordinated cooperation of transcription and replication of both the nuclear and the mitochondrial genomes. The master regulator that provides the transcriptional control of mitochondrial biogenesis is the peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α (PGC-1α) [45]. The expression of PGC-1α can be regulated by many extra and intracellular signals mediated by CREB, AMP-activated protein kinase (AMPK), Ca2+/calmodulin-dependent protein kinase (CAMK), calcineurin, and nitric oxide (NO) [45,46,47,48,49,50]. PGC-1α coactivates a number of transcription factors, including nuclear respiratory factor 1 (NRF1), estrogen-related receptors (ERRs), and PPARs. In turn, these factors activate the expression of the nuclear genes encoding almost all mitochondrial proteins. In addition, PGC-1α was also shown to promotes the expression of two isoforms of a mitochondrial transcription specificity factor, termed TFA1M and TFB2M, and the mitochondrial DNA polymerase, which induces the transcription and replication of mitochondrial DNA (mtDNA). The role of PGC-1α in regulating cellular metabolism was described in detail in recent reviews [45,51,52].
The evidence supports that the diabetes-related suppression of energy metabolism is often associated with a decrease in the content of mitochondria and the impairment of mitochondrial biogenesis in damaged cells [53,54,55]. Indeed, the expression levels of PGC-1α, mitochondrial proteins and mRNA, as well the ratio between the number of copies of mtDNA and nuclear DNA (nDNA) are reduced in many tissues and vital organs (in particular, the cardiac and skeletal muscles, the kidneys, and the brain) of both humans and laboratory animals with diabetes (Table 1). The administration of insulin to animals with type I diabetes leads to an increase in the expression level of mitochondrial proteins and an improvement of energy metabolism (an increase in the rate of ATP synthesis) [56,57]. The use of antidiabetic drugs (metformin, thiazolidinediones, empagliflozin, and some others) or regular exercise recovers the expression of PGC-1α, the mtDNA/nDNA ratio and improves mitochondrial energy metabolism in diabetic animals [58,59,60,61,62]. At the same time, an increase in the expression of PGC-1α results in the restoration of both energy functions and insulin sensitivity of cells [62,63].
Interestingly, the expression level of PGC-1α in hepatocytes can increase in diabetic conditions, leading to an elevation of the production of glucose in the liver and the aggravation of hyperglycemia [19]. The knockdown of PGC-1α in the liver tissue improves glucose tolerance and hepatic insulin sensitivity in db/db mice [64].
Meanwhile, some studies have shown that the number of mitochondria in some cells does not change or even increases in the course of diabetes. It was found that in patients with type I and type II diabetes, there is no decrease in the number of mitochondria in the pancreas [65]. In cardiomyocytes, the mtDNA/nDNA ratio, the size and number of mitochondria upon diabetes type I and II are elevated [13]. In parallel, an increase in the production of reactive oxygen species and a suppression of the function of the mitochondrial respiratory chain are observed [66,67]. One can suggest that in these studies, adaptative changes occurred. In this case, increased mitochondrial biogenesis could be a response to mitochondrial damage due to oxidative stress. Together with the suppression of mitophagy, this may lead to an increase in the number of mitochondria (including the damaged ones) in cells of some tissues upon diabetes.
Mitophagy is the process of selective destruction of mitochondria by autophagy. The mechanism is essential for a living cell to maintain mitochondrial quality and homeostasis by removing the needless or dysfunctional mitochondria. Mitophagy also protects against apoptosis and alleviates cell damage from toxic substances [68]. The stimulation of mitophagy can occur due to the activation of sirtuin-1 (SIRT1), NAD+-dependent histone/protein deacetylase, and AMPK, which, in turn, initiates two signaling pathways containing PTEN-induced putative kinase 1 (PINK1), the E3 ubiquitin ligase Parkin (PINK1/Parkin), and NIP3-like protein X (NIX)/BNIP3. These signaling mechanisms are triggered in the cell in response to the occurrence of damaged mitochondria (for example, mitochondria with low membrane potential), which leads to the formation of autophagosomes that subsequently deliver them to lysosomes for complete destruction [69]. It is the balance between mitochondrial biogenesis and mitophagy that maintains a constant number of functionally active mitochondria in the cell. Therefore, these processes can be triggered when specific signaling pathways are activated.
The expression of PINK1, Parkin, NIX, and other autophagy-related proteins was found to increase in tissues of patients with a pre-diabetic state [20]. In rodent models of type 2 diabetes induced by a high-fat diet and low doses of streptozotocin treatment, autophagy was shown to be activated in pancreatic β-cells [21,70]. With the progression of diabetes mellitus in humans and laboratory animals, the mitochondrial-specific autophagy is suppressed in many tissues and organs (pancreas, heart, skeletal muscle, eyes) [20,22,23,24,25,28,71], which may be associated with a decrease in the activation of AMPK and SIRT1 [72,73,74]. Some studies revealed that the chronically elevated level of glucose upon type I diabetes induces the accumulation of p53 protein in the cytoplasm of β-cells of the pancreas. This protein binds to Parkin, blocks its translocation to damaged mitochondria and thereby inhibits mitophagy [75]. There is evidence that anti-diabetic drugs can enhance the autophagic clearance of damaged mitochondria, which leads to the improvement of mitochondrial energetics. It was observed that metformin operates as an agonist of AMPK (and SIRT1) and increases the expression of Parkin [25,76], whereas the inhibitors of sodium-glucose linked transporter 2 (SGLT2) activate the SIRT signaling pathway and AMPK, thereby promoting mitophagy [77,78].
Thus, one can conclude that the development and progression of diabetes and its complications in humans and animals mainly leads to the suppression of both mitochondrial biogenesis and mitophagy. As a result, the number of healthy mitochondria in the cell decreases and the proportion of the depolarized organelles increases. These changes, obviously, underlie overall mitochondrial dysfunction and abnormality throughout diabetes, which will be discussed below. At the same time, a number of studies showed the stimulation of the processes of mitochondrial biogenesis and mitophagy (Figure 1). These observations suggest the occurrence of adaptive changes in various tissues, which may prevent or delay the onset of diabetes-related complications. In this regard, further research is required to elucidate the mechanisms of changes in mitochondria at different stages in the disease progression.
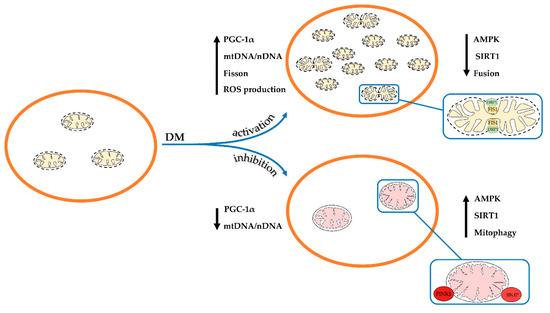
Figure 1. Schematic illustration of changes in the ultrastructure and content and of mitochondria in the cell in diabetes mellitus. Depending on the model of diabetes used or tissue, mitochondrial biogenesis and mitophagy can be enhanced or suppressed. The arrows show changes in the content of mitochondrial DNA, the main proteins responsible for mitochondrial dynamics, and the rate of ROS production by mitochondria in the pathology.
1.2. Impairment of Mitochondrial Dynamics in Diabetes Mellitus
Mitochondrial homeostasis in the cell is supported not only by the processes of mitophagy and biogenesis but also by many other mechanisms of mitochondrial quality control. Changes in the architecture of the mitochondrial network due to repeated fusion and fission of the organelles is another mechanism that regulates the maintenance of proper mitochondrial functions. Mitochondria are remarkably dynamic organelles; their shape, position in the cell, arrangement of the cristae, and other morphological signs can vary significantly depending on physiological or pathological conditions. It has been suggested that conditions that require increased energy expenditure (fasting, acute stress, and others) promote mitochondrial fusion and the formation of an interconnected/elongated mitochondrial network. Conversely, conditions of excess energy sources and relatively low demand for ATP stimulate mitochondrial division and fragmentation of the mitochondrial network, which results in an increase in the proportion of single mitochondria in the cell. In addition, mitochondrial fragmentation has been thought to be essential for mitophagy induction [79,80].
The dynamic balance between fission and fusion is regulated by large GTPase family proteins belonging to the Dynamin superfamily. Mitochondrial fission is mainly ensured by dynamin-related protein 1 (DRP1) and fission protein 1 (FIS1) [79,81]. DRP1 mediates mitochondrial construction by assembling into an oligomeric ring in the constriction sites, which divides the mitochondrion in a GTP-dependent process. Drp1 is primarily localized in the cytoplasm, but it can be recruited to mitochondria via receptors anchored into the outer membrane of the organelles. FIS1, a small adaptor protein located in the outer mitochondrial membrane, participates in the recruitment of DRP1 through its cytosolic domain. Along with FIS1 and DRP1, three more proteins have been identified to be involved in mitochondrial fission: mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51, respectively) and mitochondrial fission factor (MFF) [82].
The fusion of the outer mitochondrial membranes is mediated by mitofusins 1 and 2 (MFN1 and MFN2) [79,81]. Mfn2 is also one of the linker proteins that is involved in the formation of contact sites between mitochondria and the endoplasmic reticulum (ER), termed mitochondria-associated ER membranes (MAMs). MAMs play a fundamental role in the regulation of cellular metabolism and, in particular, glucose and Ca2+ homeostasis [83,84]. The fusion of the inner mitochondrial membranes is carried out by optic atrophy 1 (OPA1) (see reviews [79,81,85]). OPA1 has a dual role: it is also involved in maintaining the structure of mitochondrial cristae and thereby can modulate mitochondrial function through cristae remodeling [86].
There is increasing evidence that altered insulin signaling in cells may contribute to changes in the level of proteins responsible for mitochondrial fusion and in the structure of the mitochondrial network. It has been shown that the expression of MFN2and OPA1 is significantly decreased in many tissues of diabetic animals [29,30,32,33,36,87,88,89]. Treatment with the mitochondrial fusion promoter-M1 increases the expression of OPA1, promotes mitochondrial fusion, enhances mitochondrial respiratory capacity, and reduces the rate of ROS production by mitochondria [90]. At the same time, the function of the mitochondrial fission machinery in diabetes is enhanced. In particular, increased expression of DRP1 and FIS1 is observed [28,30,31,32,33,34,87,89]. In cultured skeletal muscle cells and cortical neurons, genetic and pharmacological inhibition of Drp1 was shown to attenuate palmitate-induced mitochondrial fragmentation and insulin resistance [91,92]. These events were accompanied by a decline in ROS generation by mitochondria and an increase in the mitochondrial respiratory capacity.
As mentioned above, mitochondrial fragmentation can play an important role in the induction of mitophagy in the cell. At the same time, there is evidence that DRP1 is not essential for mitophagy, but rather restricts PINK1–Parkin activity to specific mitochondrial subdomains. In this regard, one can assume that an increase in DRP1 in diabetes can lead to suppression of mitophagy [93].
Along with the enhanced fission and fragmentation of mitochondria in diabetes mellitus, pronounced alterations in the ultrastructure of the organelles are observed. Transmission electron microscopy analysis reveals the occurrence of swollen “hypertrophic” mitochondria with a decreased matrix density and disorganized inner-membrane cristae in cells from various tissues and organs [29,88].
It was found that antidiabetic drugs can restore mitochondrial fusion and fission within the cells. Notably, the SGLT2 inhibitors empagliflozin and dapagliflozin modulate the activity of mitochondrial dynamics via the regulation of fission (FIS1 and DRP1) and fusion (MFN1 and MFN2) proteins [34,94,95]. Metformin reduces the expression of DRP1 [96]. It was also demonstrated that exercise promotes a decrease in DRP1 expression and an increase in MFN1 and MFN2 expression. The dipeptidyl peptidase-4 inhibitor vildagliptin suppresses the expression of FIS1 and DRP1 and prevents the translocation of DRP1 into mitochondria [97].
Thus, one can conclude that during diabetes mellitus, the system of mitochondrial fission and fusion is thrown out of balance, which leads to the remodeling of the mitochondrial network in the cell. Ultimately, this is accompanied by a change in the structure of mitochondria and their functional activity (Figure 1).
1.3. Diabetes-Induced Changes in the Functional Activity of Mitochondria
In the course of both type 1 and 2 diabetes mellitus, abnormal glucose utilization leads to a “switch” of energy metabolism: the cells begin to replenish their energy needs mainly via fatty acid β-oxidation. Although the mechanisms by which this occurs are different for the two types of diabetes, there is eventually an increased uptake and utilization of fatty acids in both cases. An increase in the plasma level of free fatty acids and their uptake by cells in type II diabetes further reduces the insulin-dependent absorption of glucose. In parallel, high rate of gluconeogenesis is observed [98,99]. Ultimately, this results in the fact that the synthesis of ATP in cells occurs mainly due to the oxidation of fatty acids, and not the metabolism of carbohydrates [54].
In parallel, there is a PPARα-mediated increase in the expression of mitochondrial enzymes responsible for the metabolism of fatty acids, notably carnitine palmitoyl transferase 1 (CPT1) [100]. Inhibition of CPT1 is considered as a possible mechanism to modulate altered energy metabolism and improve insulin sensitivity of cells upon type II diabetes [101].
As is known, type II diabetes leads to serious disorders of protein metabolism. In mitochondria, the inhibition of pyruvate dehydrogenase, branched-chain a-ketoacid dehydrogenase, and tyrosine aminotransferase occurs. These events would selectively increase tissue and blood concentrations of some essential amino acids, particularly methionine [102]. In this regard, methionine restriction can improve insulin resistance and glucose homeostasis in diabetes [103].
As would be expected, changes in the cellular energy metabolism and structure of mitochondria in type I and II diabetes, should lead to impaired mitochondrial respiration and oxidative phosphorylation. Meanwhile, the literature data are rather contradictory.
On the one hand, some studies demonstrated that respiratory function and ATP synthesis in diabetic mitochondria are suppressed. It was found that the ADP/O ratio and the rate of ADP-stimulated respiration of isolated mitochondria harvested from diabetic patients and different animal models for diabetes significantly decrease [104,105,106]. These events are accompanied by an increase in the NADH/NAD ratio due to a pronounced dysfunction of complex I of the mitochondrial respiratory chain, as well as a decrease in the activity of complexes II, IV, and V (ATP synthase) [13,107]. The content of the ATP synthase complex in mitochondria of the liver and the heart of animals with type 1 diabetes is also reduced, which can be associated with the accumulation of calpain 1 in the organelles [41,108]. The alterations in activities and protein expression of the respiratory chain complexes both inhibit ATP synthesis and accelerate the generation of ROS in mitochondria, resulting in the development of oxidative stress. Inhibition of calpain 1 was shown to restore the levels of ATP synthase-α (ATP5A1) and autophagy, as well as prevent mitochondrial fragmentation and excessive ROS production [108,109].
It is known that the individual complexes of the mitochondrial respiratory chain are capable of organizing into multienzyme assemblies, so-called supercomplexes, which increases the efficiency of ATP synthesis. Destabilization of the structure of the mitochondrial supercomplexes might contribute to the disruption of mitochondrial respiration and oxidative phosphorylation. In tissues of patients with type 2 diabetes, the extent of assembly of the respiratory supercomplex I-III-IV was found to decrease compared with that in the control [110]. Similar changes were demonstrated in diabetic mice that were fed with a high cholesterol diet. In these animals, the rate of mitochondrial state 3 respiration, the value of the mitochondrial membrane potential, and the assembly of the respiratory supercomplexes in mitochondria of the liver were reduced [111]. Some studies also revealed that a diabetes-induced decline in the activity of complexes of the respiratory chain can be associated with a decreased content or altered composition (peroxidation) of cardiolipin, a main anionic phospholipid in the inner mitochondrial membrane (see review [54]).
On the other hand, several studies showed that diabetes is not accompanied by a significant decrease in the ability of the mitochondria to synthesize ATP. The studies were performed on mitochondria isolated from various tissues of patients with both type 1 and type 2 diabetes and some diet-induced and transgenic diabetic animal models (see review [37]). Mitochondria respiration in these experiments was supported by oxidation of substrates of complexes I and II of the respiratory chain [112,113]. In some cases, a diabetes-induced stimulation of respiration of mitochondria was even observed. For example, mitochondrial respiration was found to increase in the diabetic liver, where the level of cardiolipin in the mitochondrial membranes was also elevated [113]. Therefore, an increase in the phospholipid level in diabetes is likely to be required for adaptive responses, whereas a loss of its content in mitochondria can contribute to the dysfunction of the organelles [114].
It should be noted that some antidiabetic drugs can suppress oxidative phosphorylation of isolated mitochondria [115]. Metformin and pioglitazone inhibit the activity of complex I of the mitochondrial respiratory chain [115,116,117]. Pioglitazone at high concentrations induces mitochondrial swelling, increases ROS production, and decreases the membrane potential of mitochondria [118].
As mentioned above, the development of diabetes is accompanied by the accumulation of free fatty acids inside cells and intracellular membranes. In mitochondria, this can lead to uncoupling of respiration and oxidative phosphorylation as well as decreased ATP production [66]. The uncoupling action of free fatty acids on oxidative phosphorylation is carried out by the protonophore mechanism and mediated by anion carriers of the inner mitochondrial membrane: uncoupling proteins (UCP1-3), adenine nucleotide translocator (ANT), aspartate/glutamate carrier, etc. Despite the seemingly negative events, uncoupling of oxidative phosphorylation in mitochondria is of great physiological significance. Especially, mitochondrial uncoupling triggers nonshivering thermogenesis in brown adipose tissue and reduces the excessive generation of ROS by mitochondria [66,119,120].
Some studies reported that the expression of uncoupling proteins (UCPs) 2 and 3, which mediate proton leak across the inner mitochondrial membrane when activated by fatty acids, is increased in many tissues of animals with experimental diabetes [66,121,122,123]. Further investigations in this field revealed that UCPs may be involved in the development of mitochondrial dysfunction in diabetes, and their impact on mitochondrial metabolism is dependent on the used model of diabetes mellitus. It was found that after a long-term high-fat diet in animals, UCP3 induces mitochondrial uncoupling and reduces cardiac efficiency. Meanwhile, UCP3 does not mediate mitochondrial uncoupling in leptin-deficient states of animals (ob/ob mice). It should be noted that in the latter case, an increase in UCP expression may be an adaptive mechanism in response to an excessive ROS generation by mitochondria [124]. Studies suggested that it is the prevention of ROS overproduction and ROS- induced cell death that is the main result of an increase in the content of this protein in the mitochondrial membrane. So, overexpression of UCP2 was found to inhibit ROS generation and high glucose-induced apoptosis of human umbilical vein endothelial cells [122].
1.4. Oxidative Stress and Diabetes Mellitus
Oxidative stress is a key component in the pathogenesis of diabetes mellitus and the only pathogenic factor that almost all available studies point to [13,54,125]. Only a small number of studies indicate that mitochondrial dysfunction in diabetes is not necessarily linked with an overproduction of ROS [126,127]. Except for a few papers, most studies demonstrate that the generation of ROS by mitochondria is increased dramatically in many tissues and metabolically active organs in obesity and insulin resistance [17,20,34,38,39,40,41]. Moreover, mitochondrial ROS modulates various pathophysiological processes in cells upon diabetes, ranging from an adaptive metabolic response to ROS production and ending with ROS-induced cell death [54,125].
The main resource of ROS in mitochondria is the electron transport chain, namely, complexes I (NADH-ubiquinone oxidoreductase) and III (ubiquinol-cytochrome c oxidoreductase). In these complexes, molecular oxygen is reduced to superoxide anion radical (O2−). Superoxide can also be formed during reverse electron transfer from ubiquinol to complex I in an over-reduced electron transport chain, which leads to the reduction of NAD to NADH. Being short-lived, superoxide can spontaneously or enzymatically convert to H2O2. In turn, H2O2 and O2− can be further converted to the extremely active hydroxyl radical (OH) in the Fenton reaction. Maintaining a low level of harmful reactive radicals in cells is provided by the antioxidant defense system, which includes the enzymatic proteins manganese superoxide dismutase (MnSOD), catalase, glutathione peroxidase, thioredoxins, peroxiredoxins, as well as non-enzymatic scavengers such as glutathione, tocopherol, and others (see reviews [128,129]).
The association of an overproduction of ROS by mitochondria with the occurrence of cell resistance to insulin has been proven convincingly. This perspective was detailed in the review by Yaribeiga and others who considered five main molecular mechanisms through which oxidative stress induces insulin resistance: β-cell dysfunction, decreased expression of the glucose transporter GLUT4, suppression of insulin signaling pathways, increased inflammatory responses, and mitochondrial dysfunction [125].
It is commonly accepted that elevated ROS levels can suppress ATP synthesis in mitochondria. First, ROS can react with the unsaturated fatty acids of lipids of mitochondrial membranes and induce lipid peroxidation. The oxidative degradation of membrane phospholipids may promote non-specific permeabilization of the inner mitochondrial membrane, increase proton leakage, and inhibit the activity of respiratory chain complexes. As mentioned above, the peroxidation of cardiolipin results in the dysfunction of complex I and destabilization of supercomplex assemblies in the mitochondrial membranes during diabetes [54]. Second, suppression of oxidative phosphorylation by ROS may be associated with oxidative damage or decreased level of mitochondrial sirtuin 3 (SIRT3), a major NAD-dependent protein deacetylase [107,130]. In healthy cells, SIRT3 plays a key role in deacetylating and modifying the enzymatic activities of several mitochondrial proteins including the respiratory chain complexes I, II, and III [130]. It was shown that SIRT3 protein level is significantly reduced in the tissues of mice with type 1 and 2 diabetes [131]. In Sirt3 knock-out mice, the hyperacetylation of the mitochondrial respiratory chain complexes I and III is associated with the development of oxidative stress in cells [22,107,130]. Furthermore, these events are accompanied by the suppression of mitochondrial respiration and inactivation of the primary antioxidant enzyme MnSOD [130,132]. Third, elevated ROS formation is one mechanism of upregulation of UCPs, which eventually may lead to a decrease in mitochondria-generated ATP levels in cells [54]. Fourth, ROS-induced oxidative damage to mitochondrial proteins can disturb their functions and trigger the opening of the Ca2+-dependent mitochondrial permeability transition (MPT) pore. The pathological phenomenon of the MPT pore opening is characterized by a sudden loss of permeability of the inner mitochondrial membrane, which can lead to the collapse of the membrane potential, release of proapoptotic proteins from mitochondria, and ultimately cell death [133]. Finally, excessive ROS production can induce oxidative damage to mtDNA followed by deficiencies of the respiratory chain complexes and total mitochondrial dysfunction [134].
Interestingly, ROS can be an activator of autophagy/mitophagy [135]. It can be suggested that, like upregulation of mitochondrial uncoupling proteins, this mechanism works in pre-diabetic or early stages of diabetes mellitus. However, in the late stages of diabetes, when mitophagy is suppressed, this compensatory mechanism is apparently suppressed [20].
Recent studies suggest that treatment with metformin, thiazolidinediones, SGLT2 and DPP4 inhibitors prevents both the development of oxidative stress and mitochondrial dysfunction in diabetes [34,96,97,136,137,138]. A growing body of evidence suggests that hypoglycemic drugs eliminate most of the signs of mitochondrial dysfunction in cells of various tissues and organs of diabetic animals.
The use of regulators of oxidative stress in the comprehensive treatment of diabetes was found to maintain the structural and functional integrity of mitochondria. It has been shown that the mitochondrial-targeted antioxidants MitoTEMPO, MitoQ, BAM15, C12TPP, and CoQ10 partially prevent the ROS-induced disorders in oxidative phosphorylation and ultrastructure of mitochondria in animal models of obesity and insulin resistance [38,139,140,141,142]. Currently, the positive effects of CoQ10 and resveratrol as antidiabetic drugs have been demonstrated in most clinical trials [143]. At the same time, the administration of antioxidants has been reported to usually attenuate diabetes-associated pathological changes in cells, but not always affect glucose levels and insulin sensitivity of the organism (see review [143]).
The development and progression of oxidative stress in animal models of diabetes can also be regulated by modulating the expression and activity of major enzymes responsible for antioxidant protection. It has been shown that overexpression of catalase in muscle mitochondria of obese mice leads to the improvement of insulin sensitivity of the animals fed with a high-fat diet [144]. Increased expression of peroxiredoxin 3 or MnSOD was found to preserve cardiomyocytes and prevent the development of diabetic cardiomyopathy [145,146].
The free radical overload is one of the most common features of damaged mitochondria in diabetes mellitus. In addition to the above effects, elevated ROS levels in mitochondria dysregulates Ca2+ homeostasis through the induction of Ca2+ overload and the Ca2+-dependent formation of the MPT pore. Even though ROS are traditionally considered as one of the main activators of the mitochondrial pore, the data on the involvement of the MPT pore opening in the pathology of diabetes mellitus are currently highly controversial. In the next section, we attempt to summarize the recent data on the role of the MPT pore in the development of diabetes-related mitochondrial dysfunction and to establish the association between this pathological phenomenon in mitochondria and diabetes mellitus.
2. Ca2+Handling, MPT Pore and Diabetes Mellitus
2.1. Mitochondrial Ca2+ Transport in Diabetes
Ca2+ is a universal regulator of many intracellular processes. The appearance of Ca2+ in the cytosol of pancreatic β-cells is one of the important steps in the mechanism of insulin secretion and regulation of glucose metabolism in humans and animals [147]. On the other hand, higher intracellular Ca2+ level has been found in primary adipocytes, hepatocytes, and cardiomyocytes isolated from obese human subjects with insulin resistance as well as diabetic animals [148,149,150]. In this regard, maintaining a low Ca2+ concentration inside cells is an important function of a number of structures, including mitochondria. Activation of mitochondrial Ca2+ uptake occurs at a sufficiently high ion concentration in the cytosol. Mitochondria have the ability to rapidly transport and store high concentrations of Ca2+ in the matrix, which is extremely important for the regulation of calcium homeostasis under stressful conditions. Mitochondrial matrix Ca2+ regulates a fairly wide range of proteins of the tricarboxylic acid cycle (in particular, pyruvate dehydrogenase, citrate dehydrogenase, and α-ketoglutarate dehydrogenase), respiratory chain complexes, contributing to the maintenance of cell energy metabolism, and ATP generation. It should also be noted that excessive accumulation of Ca2+ in mitochondria leads to the opening of the MPT pore in the inner membrane and cell death initiation [84,151].Ca2+ enters the mitochondria through the voltage-dependent anion channel (VDAC) located on the outer mitochondrial membrane. VDAC is one of the components of the MAM (mitochondria-associated membrane) contacts, which allows Ca2+ released from the endoplasmic reticulum to be immediately redirected via IP3 receptors into mitochondria. The main component responsible for mitochondrial Ca2+ handling is the Ca2+ uniporter of the inner mitochondrial membrane. The uniporter has a remarkably high affinity for Ca2+, since other divalent cations are transported inside the mitochondria much more slowly and, moreover, inhibit Ca2+ uptake. It is inhibited by the polycationic dye ruthenium red and its analogues. Nowadays it is recognized that the mitochondrial calcium uniporter is a multicomponent system—the pore channel is formed by MCU integral membrane proteins (there is also an inactive MCU paralogue—MCUb), Ca2+ uptake is regulated by MICU (MICU1–3) gate proteins, as well as regulatory proteins EMRE and MCUR1. It is important to note that the level of these proteins varies in different tissues, and the ratio of regulatory and channel subunits determines the ability of the mitochondria of a particular tissue to absorb calcium ions. Along with the mitochondrial uniporter, other structures are considered as possible Ca2+ uptake mechanisms: the rapid mode of uptake or RaM, the mitochondrial ryanodine receptor, and the Ca2+/H+ exchanger Letm1. However, their contribution to mitochondrial Ca2+ uptake is not as significant compared to the Ca2+ uniporter. The structures responsible for the release of Ca2+ from mitochondria include Na+/Ca2+ and H+/Ca2+ exchanges. These systems are supposed to function in different tissues: Na+/Ca2+ exchange takes place in excitable tissues, while H+/Ca2+ exchange occurs in non-excitable tissues. It has been shown that these are systems of slow release of Ca2+ from mitochondria, the rate of ion transport through them is much lower than the rate of Ca2+ entry through the Ca2+ uniporter. The carrier responsible for the Na+/Ca2+ exchange is an antiporter of the inner mitochondrial membrane, capable of releasing Ca2+ in exchange for Na+ or Li+ (NCLX— Na+/Li+/Ca2+ exchanger). It is assumed that Letm1 functions as a Ca2+/2H+ exchanger. In addition to these systems of slow release of Ca2+ from mitochondria, a sharp rapid discharge of mitochondria from Ca2+ occurs by the opening of non-specific Ca2+-dependent mitochondrial pores. The balance between mitochondrial calcium entry and release is responsible for the maintenance of intracellular Ca2+ homeostasis under normal and pathological conditions. More details about mitochondrial Ca2+ transport systems are described in reviews [84,133,151,152].Intramitochondrial Ca2+ is shown to be involved in the regulation of insulin secretion in pancreatic β-cells under normal conditions [147]. The intake of glucose in β-cells leads to the accumulation of Ca2+ in the mitochondria, an increase in the concentration of ATP in the cells, and insulin secretion. Indeed, MCU knockout mice show inhibition of the first phase of insulin secretion [153]. It should also be noted that glucolipotoxicity is associated with suppression of Ca2+ transport in the mitochondria of β-cells and an increase in the level of ATP in the cytosol. However, the expression of MCU and NCLX did not change. The authors suggested that the dysregulation of Ca2+ transport in the mitochondria of β-cells under glucolipotoxicity is due to a change in the ultrastructure of organelles [154]. At the same time, palmitic acid upregulated MCU protein expression in mouse clonal β-cell MIN6 under normal glucose, but not high glucose medium. The authors suggested that high glucose attenuates the compensatory mechanism involving MCU in palmitate-induced cytotoxicity and causes further serious consequences related to Ca2+ overload in β-cell lipotoxicity [155].One of the diabetes-related pathological changes in β-cells is the development of ER stress [156]. Along with the suppression of Ca2+ transport in mitochondria, this causes an increase in free Ca2+ in the cytoplasm, which, in turn, can lead to an imbalance of diverse Ca2+-dependent signaling pathways. In addition, the induction of mitochondrial dysfunction and ER stress in the pancreas may eventually result in the death of β-cells and an increase in diabetic complication rates.Data on the functioning of the calcium uniporter in diabetic tissues are quite contradictory (Figure 2). A decrease in the rate of Ca2+ transport was observed in heart mitochondria in streptozotocin- induced T1DM rats and in T2DM ob/ob mice, as well as in pancreatic cells [154,157,158,159]. Heart mitochondria also showed a decrease in MCU expression in a murine model of streptozotocin-induced T1DM, which was accompanied by a suppression of mitochondrial Ca2+ uptake [160]. An elevated level of the dominant negative MCUb subunit of the uniporter is also expected to contribute to this picture. Indeed, normalization of the MCU level in hearts restored mitochondrial Ca2+ handling, increased pyruvate dehydrogenase activity, and reprogrammed a metabolism toward normal glucose oxidation [160,161]. In addition, the heart of db/db mice showed reduced expression of the peripheral membrane MiCU1 protein acting as a gatekeeper. The reconstitution of MiCU1 in diabetic hearts significantly inhibited the development of diabetic cardiomyopathy by increasing mitochondrial Ca2+ uptake and subsequently activating the antioxidant system [162].
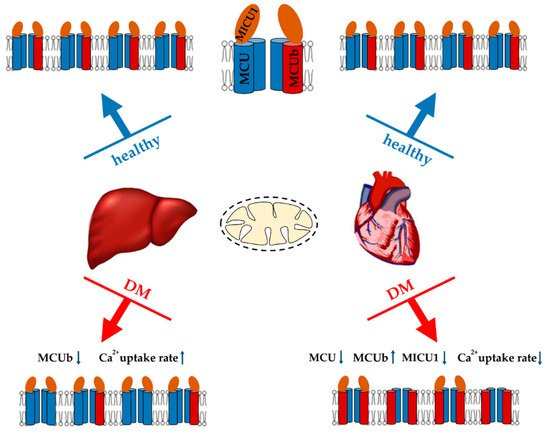
Figure 2. Major changes in the Ca2+ uniport system of liver and heart mitochondria in streptozotocin-induced diabetes mellitus (T1DM) and their effect on the Ca2+ uptake by organelles. An increase or decrease in the level of MCUC subunits, as well as in Ca2+ uptake rate is indicated by arrows (↑ or ↓).
On the other hand, there is evidence that the development of diabetes is accompanied by activation of the calcium uniporter. Indeed, as far back as the 70s, it was shown that the induction of alloxan diabetes stimulates the entry of Ca2+ into liver mitochondria [163]. Recently, we also demonstrated that two weeks after streptozotocin administration to Sprague-Dawley rats, the rate of Ca2+ uptake by liver mitochondria significantly increased. The analysis showed that an increase in the Ca2+ transport rate was due to a decrease in the expression of the dominant-negative MCUb subunit of the uniporter [41]. Adipose tissue mitochondria also show an increase in Ca2+ handling. It was shown that mitochondrial Ca2+ uptake increased and MCU components (MCU and MICU1) were upregulated in insulin-resistant adipocytes. Similar results were observed in mouse (db/db and ob/ob) and human visceral adipose tissue during the progression of obesity and diabetes [164].The development of diabetes mellitus changes not only the activity and expression of Ca2+ uniporter, but also NCLX. Indeed, the endothelia of streptozotocin-induced T1DM rats demonstrated an increase in NCLX expression. In this case, silencing of NCLX expression increased ROS generation and NLRP3 inflammasome activation [165].Insulin resistance and T2DM cause a disruption in the structure of MAM contacts [157,166,167,168]. The antidiabetic drugs metformin and rosiglitazone restore the structure of MAM contacts in diabetic animals [168]. It should be noted that diabetes mellitus is associated with overexpression of VDAC1 in certain tissues (pancreatic β-cells, vascular endothelial cells) [169,170,171]. In parallel, an increased amount of Ca2+ accumulates in mitochondria, which ultimately leads to the activation of apoptosis. Inhibition of VDAC1 overexpression leads to the suppression of apoptosis in endothelial cells and improves insulin secretion in islets [170,171].The contradictions observed in studies of mitochondrial Ca2+ transport in diabetes mellitus are difficult to explain. It is possible that the development of diabetes shows tissue specificity. As mentioned above, liver cells react differently to diabetes. In particular, this organ shows PGC1-1α overexpression and stimulation of biogenesis. It is worth noting that similar adaptive changes may possibly occur in other tissues in the early stages of diabetes. Several studies on the induction of diabetes have shown an increase in the concentration of Ca2+ in the cytosol and mitochondria. In this regard, it can be speculated that under these conditions, the observed activation of Ca2+ uptake and release systems from mitochondria will lead to Ca2+ recyclization through the mitochondrial membrane (futile cycle) and ΔΨ decrease. Like UCP expression, this adaptive reaction will suppress oxidative stress, at least in the initial stages of the development of the disease. Such a futile cycle, causing a slight depolarization, is expected to stimulate mitophagy. Along with increased biogenesis, this will trigger the renewal of the mitochondrial population in the cell. Meanwhile, excessive accumulation of Ca2+ in mitochondria will undoubtedly cause the opening of the MPT pore and cell death initiation.
2.2. Mitochondrial Permeability Transition Pore
Excessive accumulation of Ca2+ in the mitochondrial matrix is known to lead to an abrupt increase in nonspecific permeability of the inner mitochondrial membrane (referred to as the mitochondrial permeability transition (MPT) pore) for various ions and hydrophilic compounds with a molecular weight of up to 1.5 kDa. This leads to swelling of the mitochondria, equilibration of ionic gradients across the inner membrane, a decrease in the mitochondrial membrane potential, and impaired ATP synthesis. The final consequence of the opening of the MPT pore is cell death. Ca2+-dependent permeabilization of the inner mitochondrial membrane is one of the key elements in the process of cell death during hypoxia and subsequent reoxygenation. Moreover, convincing evidence has accumulated over the years supporting an essential role of the MPT pore opening in the development of cardiovascular diseases, neurodegenerative processes, viral diseases, muscular dystrophies, etc. [84,172,173,174,175].By the mid-90s of the last century, most of the modulators of the MPT pore had been elucidated. This is described in detail in a review by Zoratti and Szabo. Ca2+ is perhaps the main pore activator. In addition to Ca2+, inorganic phosphate (and polyphosphates), SH-oxidizing agents, oxidative stress, uncouplers, a decrease of the mitochondrial adenine nucleotide content, and other factors stimulate MPT pore opening. Inhibitors of the mitochondrial pore are the cyclosporins (cyclosporin A is most effective), adenine nucleotides, SH-reducing agents, reduced pyridine nucleotides, etc. [175].Despite significant progress in the study of MPT pore induction and regulation, its molecular structure and protein composition are still under discussion (Figure 3). An analysis of the literature data suggests that the MPT pore is a nonselective, high-conductance megachannel consisting of proteins of the inner and outer mitochondrial membranes. However, to date, cyclophilin D is the only, precisely established component of this structure; it is the pharmacological target of cyclosporin A and its analogues, which can specifically block pore opening [176,177]. Cyclophilin D is considered as a regulatory protein, which in the presence of Ca2+ stimulates rearrangement in the proteins responsible for the formation of the MPT pore channel. Knockout of cyclophilin D or its binding to an inhibitor leads to a significant increase in the threshold concentration of Ca2+ necessary for the pore opening. In 2015, using RNAi-based screening, it was suggested that, along with cyclophilin D, spastic paraplegia 7 (SPG7) is an important regulatory component of MPT [176]. However, it has recently been shown that SPG7 does not constitute a core component of MPT, but instead regulates activity by lowering the basal mitochondrial Ca2+ levels via regulation of MCUR1 and Ca2+ uniporter assembly [178].
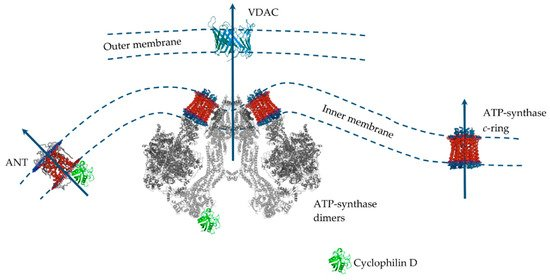
Figure 3. The putative MPT pore components of the inner and outer mitochondrial membranes. Proteins are drawn as ribbon representations (modified PDB ID codes: 6RD4 (F-ATP synthase); 5CBV (Cyclophilin D); 2JK4 (VDAC); 1OKC (ANT).
Several MPT models have been proposed over the past 40 years. Indeed, only cyclophilin D is an integral component of the pore. In this case, proteins of the outer membrane and intermembrane space (VDAC, TSPO, HK, and CrK) are auxiliary in the assembly of the pore complex in intact mitochondria [179]. However, the question of which protein is the main pore component in the inner mitochondrial membrane has not yet been resolved. Until the mid-2000s the prevailing hypothesis was that such a channel-forming pore protein of the inner mitochondrial is adenine nucleotide translocator. This assumption was because the adenine nucleotide translocator inhibitors, atractyloside and carboxyatractyloside, stimulated pore opening, and bongkrekic acid showed an inhibitory effect. Adenine nucleotides carried by the translocator under normal conditions also suppressed the pore opening [175]. In addition, it was shown that ANT is able to bind to VDAC and hexokinase, as well as to cyclophilin D, forming channels in liposomes whose properties resemble MPT [180,181,182]. However, the discovery that the opening of the MPT pore also occurred in the mitochondria from ANT1 and ANT2 null mice led to the abandonment of this model [183]. After this, the idea of a phosphate carrier as a channel component of the MPT pore was considered [184].According to the data of the last decade, mitochondrial ATP synthase is considered the main candidate for the role of the channel-forming component of the MPT pore, whose subunits and, in particular, OSCP, are able to combine with cyclophilin D, which, as expected, leads to pore opening [84,172,173,174]. In addition, it was shown that the OSCP subunit of ATP synthase contains a unique pH-sensitive histidine (H112), which has a significant modulating effect on the opening of the MPT pore [185]. Several suggestions have been made as to how ATP synthase can form an MPT pore channel. Two main hypotheses can be distinguished: (1) «dimer» and (2) «c-ring» [174]. According to the first one, ATP synthase dimers, but not monomers, conduct currents when inserted into planar lipid bilayers which are activated by Ca2+ and oxidizing agents and closed by ADP/Mg2+ (established MPT pore desensitizers) [186]. Further, it was shown that currents were strongly attenuated in yeast mutants that lacked ATP synthase subunits e and g necessary for the formation of dimers [187]. Indeed, channel formation by ATP synthase dimers was shown on mitochondria of evolutionarily distant species (S. cerevisiae, and D. Melanogaster), which makes this hypothesis quite convincing [187,188,189]. At the same time, it was found that the ATP synthase monomer is sufficient, and dimer formation is not required, for MPT pore activity [190]. According to an alternative «c-ring» hypothesis, the c-subunit of ATP synthase localized in the inner membrane of organelles may act as a channel component of the MPT pore [191,192]. In this case, it is assumed that Ca2+ and (or) ROS-induced dissociation of the F1 sector of ATP synthase leads to conformational changes in the c-ring of the Fo sector, which allows the formation of an MPT pore channel. However, this hypothesis runs into a series of contradictions. Indeed, in this case, the process of dissociation must be fast and reversible. First, there must be a quick detachment of the F1 sector, and second, the c-ring channel must be emptied and hydrated, which makes this hypothesis unlikely [174,193]. Moreover, the data of model experiments suggest that the hypothetical MPT pore based on the c-ring will have a significantly lower conductivity than that shown for MPT [194]. These data are supported by the results of patch-clamp measurements. Finally, it has recently been shown that mitochondria of mutant cells with disrupted c-subunits of ATP synthase still display a cyclosporin A-sensitive Ca2+-induced MPT pore opening and, moreover, in some cases demonstrate increased sensitivity to the induction of this process [195].Recent data again bring us back to the question of ANT as a structural element of the MPT pore. Indeed, it has recently been shown that the MPT pore in c-subunit-deficient mitochondria is sensitive to cyclosporin A, ADP, and bongkrekic acid [195]. Finally, knockout of the genes of three ANT isoforms (not two, as in 2005) significantly increased mitochondrial resistance to MPT induction and the calcium capacity of organelles, nevertheless, did not prevent it [196]. These contradictions led to the emergence of a model for the joint participation of ANT and ATP-synthase in the MPT pore opening. It is assumed that, depending on the threshold concentration of Ca2+, these proteins will form channels of various currents that provide different modes of MPT functioning [197] and contribute to both the rapid release of Ca2+ and metabolites and the maintenance of the functioning of the futile cycles mentioned above.It should also be mentioned that the search for MPT modulators made it possible to establish conditions when the mitochondrial Ca2+-dependent swelling was insensitive to cyclosporin A. This type of permeabilization includes the lipid pore induced by saturated fatty acids and Ca2+. In our previous papers, we described this type of permeabilization of the mitochondrial membrane in sufficient detail [84,198,199]. We found that the opening of this pore occurs by the mechanism of a chemotropic phase transition in a lipid bilayer. Indeed, palmitic acid in the presence of Ca2+ was able to permeabilize both natural and artificial membranes. The physiological significance of this pore has also been described [200]. The features of the formation and physiological significance of cyclosporin A-insensitive lipid pore are presented in more detail in our previous review [84].
2.3. MPT Pore as a Target for Diabetes Management
The issue of MPT pore as a target in the treatment of diabetes mellitus is rather complicated. According to a wide range of studies, cyclosporin A and other MPT pore inhibitors contribute to the suppression of mitochondrial dysfunction and improve the quality of life of animals [217,219,232]. Administration of high-dose cyclosporin A has been demonstrated to induce remission of type 1 diabetes mellitus [240]. On the other hand, the same cyclosporin causes suppression of mitochondrial biogenesis in liver cells [220]. Mice with the deletion of cyclophilin D show the development of hyperglycemia, insulin resistance, and glucose intolerance, albeit resistant to diet-induced obesity [241]. However, it should be noted that the in vivo interpretation of the effects of cyclosporin A as an MPT inhibitor may not always be correct. Cyclosporin A is a well-known immune suppressor, and its effect can be associated not only with the suppression of MPT, but also with the effect on various signaling pathways in humans and animals.Synthetic and natural antidiabetic compounds exhibit a bi-directional effect on MPT pore opening. Notably, metformin has been shown to inhibit MPT pore opening in mitochondria, enhances biogenesis, and prevents cell death [61,242,243]. However, there is an evidence that metformin stimulates MPT in rat liver mitochondria [244]. The thiazolidinedione class of antidiabetic agents also shows a similar stimulating effect on MPT pore [245]. Moreover, troglitazone enhanced MPT pore induction in the liver of diabetic Zucker (fa/fa) rats [246]. The natural polyphenolic compound luteolin, reduces mortality from coronary artery diseases, including diabetes [107]. It has been shown to inhibit MPT pore opening [247]. On the other hand, the plant alkaloid berberine, used in traditional Chinese medicine and possessing antidiabetic properties [248], causes an inhibition of mitochondrial respiration and a decrease on Ca2+ loading capacity through induction of the mitochondrial permeability transition [249]. All this suggests that it is necessary to carefully approach the issue of diabetes mellitus therapy through the modulation of MPT pore activity.
3. Conclusions
Management of mitochondrial dysfunction can undoubtedly contribute to improving glucose metabolism and seems promising in the context of diabetes therapy. At the same time, it must be remembered that diabetes affects the functioning of mitochondria in various organs and tissues with different intensities. This is clearly seen especially in the MPT pore opening in mitochondria. The use of a wide range of animal models, regimens, and duration of exposure to diabetic stress often leads to rather contradictory results regarding mitochondrial functional changes in diabetes. It is clear that mitochondria play a key role in both compensatory processes and pathological changes under diabetic stress. In this regard, it is necessary to carefully approach the issue of regulation of mitochondrial dysfunction in diabetes mellitus and conduct detailed comprehensive all organ studies of compounds that affect the functioning of mitochondria.

