Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Miriam Longo | + 2110 word(s) | 2110 | 2022-03-14 10:23:19 | | | |
| 2 | Camila Xu | + 3 word(s) | 2113 | 2022-03-15 04:33:58 | | | | |
| 3 | Camila Xu | + 3 word(s) | 2113 | 2022-03-15 04:34:59 | | | | |
| 4 | Camila Xu | Meta information modification | 2113 | 2022-03-15 04:35:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Longo, M.; Meroni, M.; Dongiovanni, P.; Paolini, E. Acute Intermittent Porphyria. Encyclopedia. Available online: https://encyclopedia.pub/entry/20544 (accessed on 23 July 2026).
Longo M, Meroni M, Dongiovanni P, Paolini E. Acute Intermittent Porphyria. Encyclopedia. Available at: https://encyclopedia.pub/entry/20544. Accessed July 23, 2026.
Longo, Miriam, Marica Meroni, Paola Dongiovanni, Erika Paolini. "Acute Intermittent Porphyria" Encyclopedia, https://encyclopedia.pub/entry/20544 (accessed July 23, 2026).
Longo, M., Meroni, M., Dongiovanni, P., & Paolini, E. (2022, March 14). Acute Intermittent Porphyria. In Encyclopedia. https://encyclopedia.pub/entry/20544
Longo, Miriam, et al. "Acute Intermittent Porphyria." Encyclopedia. Web. 14 March, 2022.
Copy Citation
Acute intermittent porphyria (AIP) is an autosomal dominant disease caused by the hepatic deficiency of porphobilinogen deaminase (PBGD) and the slowdown of heme biosynthesis. AIP symptomatology includes life-threatening, acute neurovisceral or neuropsychiatric attacks manifesting in response to precipitating factors.
AIP
PBGD
heme
liver metabolism
1. Introduction
Porphyrias are a group of rare genetic disorders caused by inborn errors in one of the eight enzymes involved in heme biosynthesis, and they lead to a reduction in heme availability alongside the overproduction and accumulation of harmful heme precursors (porphyrins). Although heme synthesis occurs in all cell types, 80% and 15% of its biogenesis arises from erythrocytes and hepatocytes, respectively [1]. Indeed, porphyrias are categorized into two different groups depending on the principal site of porphyrin accumulation: the erythropoietic cutaneous porphyrias (ECPs) and the acute hepatic porphyrias (AHPs). The latter includes δ-aminolevulinic acid (ALA) dehydratase deficiency porphyria (ADP), acute intermittent porphyria (AIP), variegate porphyria (VP), and hereditary coproporphyria (HCP) [2][3].
AIP is the most common and severe form of acute porphyria with hepatic implications, and results from a deficiency of about 50% in the hydroxymethylbilane synthase (HMBS) gene, which encodes the third enzyme of the heme pathway, referred to as porphobilinogen deaminase (PBGD). AIP’s clinical manifestations include life-threatening, acute neurovisceral and psychiatric attacks precipitated by metabolic, hormonal, and environmental factors such as fasting, infections, drugs, alcohol, or physical stress. Precipitating factors increase the demand for hepatic heme production, which normally exerts negative feedback on 5-aminolevulinic acid synthase-1 (ALAS1), the first rate-limiting enzyme of the heme pathway. Nonetheless, the deficiency in the free heme pool caused by the partial loss of PBGD activity leads to upregulation of ALAS1, thereby promoting the excess of the heme neurotoxic metabolites as the 5-aminolevulinic acid (ALA) and porphobilinogen (PBG), which accumulate in porphyric livers, plasma, and urine. The most frequent symptoms reported during the crisis are abdominal pain, nausea, vomiting, weakness, and constipation, and the crisis is often accompanied by sympathetic nervous system discomfort [4]. Peripheral motor neuropathy, complications of the central nervous system, and other chronic conditions, including hypertension, hyponatremia, and kidney disorders, may arise in patients with severe AIP, which represents a growing life-long burden for patients and their relatives [4][5][6].
Diagnosis of AIP is frequently underestimated and disease surveillance aims to educate at-risk HMBS-mutation carriers to avoid exposure to precipitating factors. Management and/or prophylaxis of acute episodes include the administration of hemin as the first-line approach and carbohydrate loading in cases of mild attacks and absence of hemin. Both therapies induce the downregulation of hepatic ALAS1, thus alleviating symptoms and reducing porphyrin levels. However, the only curative option is through the replenishment of the defective PBGD to the extent that refractory AIP recipients who underwent liver transplantation (LT) have shown a complete biochemical and clinical remission [7][8][9][10].
The major steps forward for the treatment of AIP were carried out in recent years with (1) the approval of givosiran (Alnylam Pharmaceuticals, Inc., Cambridge, MA, USA), which inhibits ALAS1 through RNA interference (RNAi) in patients aged at least 12 years, and (2) with the ongoing efforts in the development of therapies based on the replacement of nonfunctional PBGD with hepatic delivery of the mRNA or protein [11][12][13][14][15]. Interestingly, the characterization of metabolic profiles in AIP experimental models and humans has highlighted alterations in glucose and lipid metabolism, insulin resistance (IR), and hepatic mitochondrial dysfunction, which offers the possibility of considering insulin and insulin-mimetics for the clinical management of AIP [16][17][18].
2. The Link between Penetrance, Prevalence, and Genetic Traits in Acute Intermittent Porphyria (AIP)
AIP is an autosomal dominant inherited disorder with a low penetrance and significant heterogeneity of mutations in the HMBS gene [19], located on the 11q24.1–q24.2 chromosome. Currently, over 400 HMBS mutations have been recognized in the AIP scenario, which leads to the loss of PBGD enzymatic activity (Human Gene Mutation Database HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). Alternatively, spliced transcript variants encode two different isoforms of the HMBS gene: the erythroid-specific one is encoded by exons 2–15 and its promoter is positioned at intron 1, while the housekeeping isoform is encoded by exon 1 and exons 3–15 with the housekeeping promoter located in the 5’ flanking region upstream of exon 1 [20]. The majority of patients with AIP carries the HMBS mutations in exons 3–15, which affects both PBGD isoforms, while mutations in exon 1 do not influence PBGD functionality in erythrocytes [20]. Additionally, defects in the 5′-promoter region of the HMBS gene have been detected in individuals with AIP. Regarding the 400 mutations in the HMBS gene, there are 31 CpG dinucleotides in the 1086 base-pair coding sequence that are considered mutable as a consequence of the oxidative deamination of methylated cytosines [21][22]. Although AIP is a low-penetrance disorder, it has been identified that few HMBS mutations are relatively common, such as the p.R173W and p.R167Q variants, caused by CpG methylation, and the p.G111R and p.W198X variants, which are most frequent in Argentina and Sweden due to the founder effect [23][24][25].
To unravel the phenotypic expression and transmission of AIP, Lenglet and colleagues exploited the Exome Variant Server (EVS) database to estimate the prevalence and penetrance of deleterious HMBS mutations in the general population and performed an intra-familial study that included 253 families from the French reference center for porphyria (CFP). Although the prevalence of AIP genetic traits was high (1/1299), they estimated its penetrance at 0.5–1% in the general population. Conversely, in French families, they identified 496 subjects who were symptomatic, and 1672 relatives who were asymptomatic HMBS carriers, rating the penetrance of AIP at 22.9%. Moreover, the EVS database recognized 127 HMBS nucleotide variants from subjects of Caucasian and African families. These genetic variations included 22 heterozygous missense mutations, 12 synonymous mutations, 11 untranslated region (UTR) variants, and 82 intron variants, among which 20 missense mutations appeared to be harmful in the descendants of French patients with AIP. Particularly, the p.Ala122Pro, p.Arg167Gln, p.Arg175Gln, p.Arg195Cys, and p.Arg355Gln variants encoded an inactive protein, while the p.Glu86Val, p.Arg321His, and p.Asp359Asn mutations reduced PBGD enzymatic activity to 50%. The presence of null alleles, leading to the complete loss of function of the PBGD protein, was more frequent than missense mutations in families with AIP and correlated with the severity of disease manifestation [26]. An intrafamilial study identified a significant phenotypic correlation—defined as the manifestation of a symptomatic trait—among siblings, which may be explained by genetics, with a probability of 73%. This correlation decreased from first to third degree relatives, suggesting that the outcome of the disease was even influenced by other genetic factors. In addition, the phenotypic correlation was higher between kinship members of the same age and decreased between different generations, indicating that even environmental factors could modulate the risk of acute episodes and influence AIP inheritance [26]. Accordingly, a 3-year prospective study of symptomatic patients with AIP in 11 European countries revealed that the annual incidence of symptomatic manifestations was around 0.13 per million, while its prevalence was 5.9 per million in Europe [27]. The discrepancy in the prevalence of HMBS mutations and the occurrence of acute manifestations, coupled with an estimated penetrance of 20–50% in families with AIP versus 1% for the general population, strongly emphasize the role of environmental factors in triggering AIP attacks [26][27].
As we previously mentioned, AIP is an autosomal dominant disorder with the same mutational distribution among men and women, even if the manifestation of acute attacks affects more females than males. Accordingly, Baumann and colleagues found that in Finland, women displayed an AIP penetrance of 41% [28].
The majority of HMBS mutations encompassing p.W198X, c.1073delA, and p.R26C were correlated with both higher penetrance and clinical manifestations, while other variants such as p.R167W, p.R225G, and c.G33T were associated with lower penetrance and mild clinical events. Concerning the p.R173W mutation, a reduced penetrance was observed in Finland, whereas it was much higher in Northern Sweden and Spain [29]. A correlation between the presence of p.R116W, p.R173W, p.R149X, p.Q217H, p.G218R, p.A219P, and p.A330P mutations and the severity of AIP manifestations was also predicted through a bio-informatics approach [30]. To conclude, AIP prevalence in the population is high, although its penetrance is exceptionally low, resulting in the difficult identification of asymptomatic individuals with AIP. Moreover, the different penetrance rates of symptomatology in the relatives of French patients with AIP underline that AIP inheritance could be modulated by the environment and other genetic factors independently of HMBS variations.
3. Heme Biosynthesis and AIP Pathogenesis
The first step in the heme pathway begins in the mitochondria, where glycine and succinyl-coenzyme A (succinyl-coA), derived from the Krebs cycle, are converted into ALA through the ALAS enzyme [2]. ALAS exists in isoforms which are encoded by two separate genes: the ubiquitously expressed ALAS1, and the erythroid-specific isoform ALAS2. Two molecules of ALA are transported into the cytoplasm and are condensed by ALA dehydratase (ALAD), producing monopyrrole-PBG. In the third step of heme biosynthesis, four molecules of PBG are combined by PBGD enzyme to form hydroxymethylbilane (HMB), which is converted to uroporphyrinogen (UROgen) III by UROgen III synthase (UROS). Due to its molecular instability, HMBS may spontaneously form into a tetrapyrrolic ring (UROgen I) when the UROS enzyme is absent.
Then, UROgen III decarboxylase (UROD) removes the carboxyl groups from the side chains of uroporphyrinogen III to obtain coproporphyrinogen (COPROgen) III. At this step, COPROgen III is shuttled into the mitochondria, where COPROgen III oxidase (CPOX) catalyzes two sequential steps of oxidative decarboxylation, thereby forming protoporphyrinogen (PROTOgen) IX. Subsequently, PROTOgen oxidase (PPOX) removes six atoms of hydrogen from the PROTOgen IX molecule, producing protoporphyrin (PROTO) IX, the first colored tetrapyrrole intermediate. Finally, the insertion of a ferrous ion (Fe2+) into the PROTO IX ring by ferrochelatase (FECH) allows the final production of heme [31].
This molecule is incorporated into hemoproteins (e.g., cytochrome (CYP) P450 and hemoglobin), thereby participating in a wide array of intrahepatic and extrahepatic functions, including mitochondrial oxidative phosphorylation (OXPHOS), drug detoxification, and systemic oxygen transport.
AIP is caused by genetic mutations affecting PBGD activity with the consequent depletion of heme production in the liver. The main consequence of the shutdown of heme biosynthesis is the loss of negative feedback on ALAS1, which leads to the overproduction and accumulation of the porphyrin precursors ALA and PBG in the liver and systemic circulation. The excess of ALA is the major trigger of neurological damage by inducing autonomic and peripheral neuropathy and also encephalopathy. Due to its structural similarity with the γ-aminobutyric acid (GABA) and glutamate neurotransmitters, ALA may modify the GABAergic system by acting as a GABA-receptor agonist and by inhibiting GABA release from the pre-synaptic terminals [6][32]. Moreover, ALA participates in the production of free radicals and reactive oxygen species (ROS) and may promote hepatic ROS-induced genomic and mitochondrial DNA damage (mtDNA) that predisposes patients with AIP to a higher risk of developing hepatocellular carcinoma (HCC) [33][34][35]. Although it has been broadly established that the symptoms of acute attacks are mainly attributed to the accumulation of neurotoxic metabolites, there is no conclusive evidence to date about the factors influencing penetrance and the wide variability in clinical manifestations. Indeed, ~90% of patients with AIP, many of whom are asymptomatic, high excreters (ASHE) of porphyrins, remain asymptomatic throughout their life, suggesting that the pathophysiology of acute events may not be directly triggered by the accumulation of neurotoxic byproducts.
Metabolic alterations were observed in both animal models and patients with AIP and suggest new insights into the pivotal role of the liver in predisposing the pathogenesis of acute attacks. The first evidence was obtained from patients with AIP who improved biochemical and clinical abnormalities after LT [36][37]. Conversely, patients with HCC who received livers from donors with AIP developed neurovisceral symptoms together with increases in ALA and PBG levels [10].
Most recently, it emerged that AIP experimental models show impaired glucose metabolism and mitochondrial respiration capacity during acute attacks, while carriers of AIP displayed high body weight, hyperinsulinemia, and alterations in serum lipid profiles combined with IR [16][38][39]. These findings suggest that AIP may be considered a genetic disorder characterized by an umbrella of metabolic dysfunctions, thereby adding new additional knowledge about its pathophysiology and, possibly, novel curative strategies. The effect of PBGD haploinsufficiency on heme biosynthesis is represented in Figure 1.
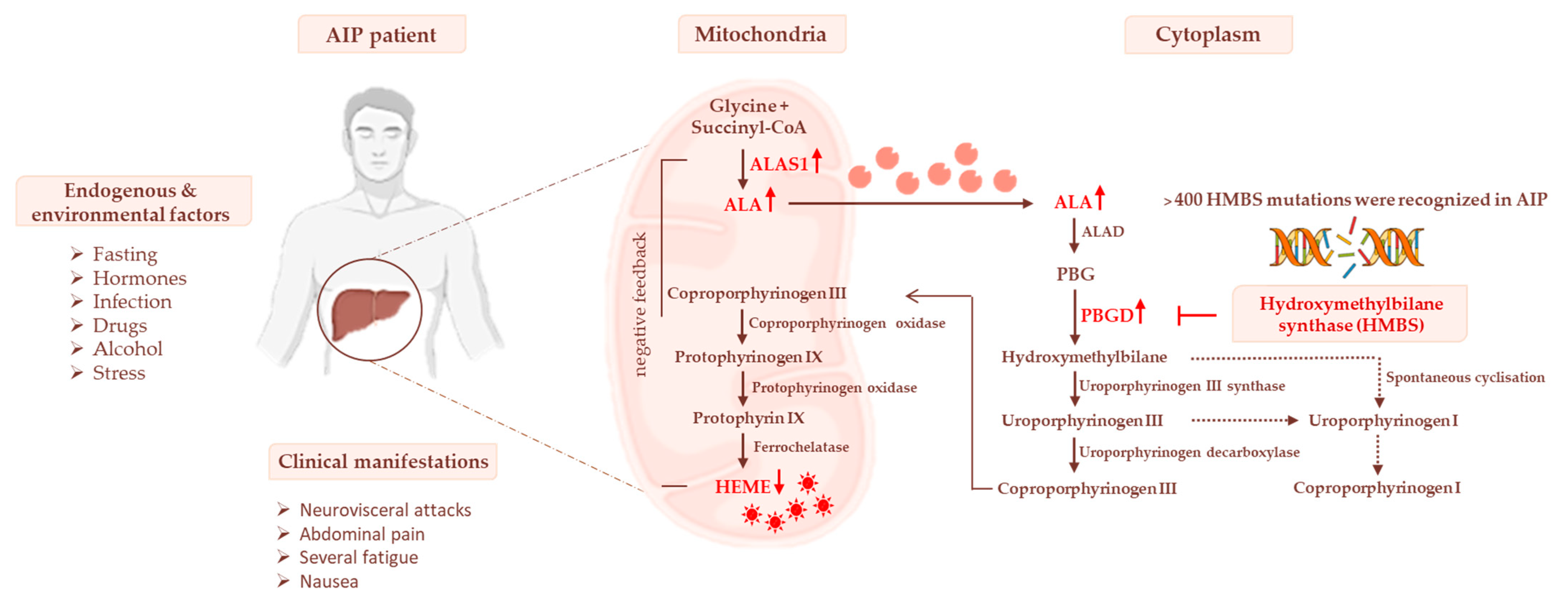 Figure 1. The heme biosynthetic pathway and AIP pathophysiology. Heme biosynthesis begins in mitochondria with the conversion of glycine and succinyl-CoA to δ-aminolevulinic acid (ALA) through the δ-aminolevulinic acid synthase-1 (ALAS1) enzyme. In the cytoplasm, ALA is metabolized to porphobilinogen (PBG) and then to COPROgen III. The latter is then transported into mitochondria for the synthesis of heme, which in turn downregulates ALAS1. The deficiency of HMBS genes causes a reduction in hepatic heme synthesis, leading to the stopping of its negative feedback on ALAS1. Thereby, the levels of porphyrin precursors (ALA and PBG), mainly in response to precipitating factors, may accumulate in the liver, systemic circulation, and urine, triggering neurological damage.
Figure 1. The heme biosynthetic pathway and AIP pathophysiology. Heme biosynthesis begins in mitochondria with the conversion of glycine and succinyl-CoA to δ-aminolevulinic acid (ALA) through the δ-aminolevulinic acid synthase-1 (ALAS1) enzyme. In the cytoplasm, ALA is metabolized to porphobilinogen (PBG) and then to COPROgen III. The latter is then transported into mitochondria for the synthesis of heme, which in turn downregulates ALAS1. The deficiency of HMBS genes causes a reduction in hepatic heme synthesis, leading to the stopping of its negative feedback on ALAS1. Thereby, the levels of porphyrin precursors (ALA and PBG), mainly in response to precipitating factors, may accumulate in the liver, systemic circulation, and urine, triggering neurological damage.References
- Balwani, M.; Wang, B.; Anderson, K.E.; Bloomer, J.R.; Bissell, D.M.; Bonkovsky, H.L.; Phillips, J.D.; Desnick, R.J. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology 2017, 66, 1314–1322.
- Stein, P.E.; Badminton, M.N.; Rees, D.C. Update review of the acute porphyrias. Br. J. Haematol. 2017, 176, 527–538.
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177.
- Bonkovsky, H.L.; Maddukuri, V.C.; Yazici, C.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.R.; Phillips, J.D.; Naik, H.; Peter, I.; Baillargeon, G.; et al. Acute porphyrias in the USA: Features of 108 subjects from porphyrias consortium. Am. J. Med. 2014, 127, 1233–1241.
- Kauppinen, R. Porphyrias. Lancet 2005, 365, 241–252.
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937.
- Frei, P.; Minder, E.I.; Corti, N.; Muellhaupt, B.; Geier, A.; Adams, H.; Dutertre, J.P.; Rudiger, A.; Dutkowski, P.; Maggiorini, M.; et al. Liver Transplantation because of Acute Liver Failure due to Heme Arginate Overdose in a Patient with Acute Intermittent Porphyria. Case Rep. Gastroenterol. 2012, 6, 190–196.
- Soonawalla, Z.F.; Orug, T.; Badminton, M.N.; Elder, G.H.; Rhodes, J.M.; Bramhall, S.R.; Elias, E. Liver transplantation as a cure for acute intermittent porphyria. Lancet 2004, 363, 705–706.
- Stein, P.; Badminton, M.; Barth, J.; Rees, D.; Stewart, M.F. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann. Clin. Biochem. 2013, 50, 217–223.
- Singal, A.K.; Parker, C.; Bowden, C.; Thapar, M.; Liu, L.; McGuire, B.M. Liver transplantation in the management of porphyria. Hepatology 2014, 60, 1082–1089.
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301.
- Honor, A.; Rudnick, S.R.; Bonkovsky, H.L. Givosiran to treat acute porphyria. Drugs Today 2021, 57, 47–59.
- Fontanellas, A.; Ávila, M.A.; Anderson, K.E.; Deybach, J.C. Current and innovative emerging therapies for porphyrias with hepatic involvement. J. Hepatol. 2019, 71, 422–433.
- Fontanellas, A.; Ávila, M.A.; Berraondo, P. Emerging therapies for acute intermittent porphyria. Expert Rev. Mol. Med. 2016, 18, e17.
- Córdoba, K.M.; Serrano-Mendioroz, I. Recombinant porphobilinogen deaminase targeted to the liver corrects enzymopenia in a mouse model of acute intermittent porphyria. Sci. Transl. Med. 2022, 14, eabc0700.
- Solares, I.; Izquierdo-Sánchez, L.; Morales-Conejo, M.; Jericó, D.; Castelbón, F.J.; Córdoba, K.M.; Sampedro, A.; Lumbreras, C.; Moreno-Aliaga, M.J.; Enríquez de Salamanca, R.; et al. High Prevalence of Insulin Resistance in Asymptomatic Patients with Acute Intermittent Porphyria and Liver-Targeted Insulin as a Novel Therapeutic Approach. Biomedicines 2021, 9, 255.
- Longo, M.; Paolini, E.; Meroni, M.; Duca, L.; Motta, I.; Fracanzani, A.L.; Di Pierro, E.; Dongiovanni, P. α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics 2021, 11, 1628.
- Vilas, G.L.; Aldonatti, C.; San Martín de Viale, L.C.; Ríos de Molina, M.C. Effect of alpha lipoic acid amide on hexachlorobenzene porphyria. Biochem. Mol. Biol. Int. 1999, 47, 815–823.
- Yasuda, M.; Chen, B.; Desnick, R.J. Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol. Genet. Metab. 2019, 128, 320–331.
- Chen, C.H.; Astrin, K.H.; Lee, G.; Anderson, K.E.; Desnick, R.J. Acute intermittent porphyria: Identification and expression of exonic mutations in the hydroxymethylbilane synthase gene. An initiation codon missense mutation in the housekeeping transcript causes “variant acute intermittent porphyria” with normal expression of the erythroid-specific enzyme. J. Clin. Investig. 1994, 94, 1927–1937.
- Brancaleoni, V.; Granata, F.; Colancecco, A.; Tavazzi, D.; Cappellini, M.D.; Di Pierro, E. Seven novel genetic mutations within the 5’UTR and the housekeeping promoter of HMBS gene responsible for the non-erythroid form of acute intermittent porphyria. Blood Cells Mol. Dis. 2012, 49, 147–151.
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988, 78, 151–155.
- Petersen, N.E.; Nissen, H.; Hørder, M.; Senz, J.; Jamani, A.; Schreiber, W.E. Mutation screening by denaturing gradient gel electrophoresis in North American patients with acute intermittent porphyria. Clin. Chem. 1998, 44 Pt 1, 1766–1768.
- De Siervi, A.; Rossetti, M.V.; Parera, V.E.; Astrin, K.H.; Aizencang, G.I.; Glass, I.A.; Batlle, A.M.; Desnick, R.J. Identification and characterization of hydroxymethylbilane synthase mutations causing acute intermittent porphyria: Evidence for an ancestral founder of the common G111R mutation. Am. J. Med. Genet. 1999, 86, 366–375.
- Ma, L.; Tian, Y.; Peng, C.; Zhang, Y.; Zhang, S. Recent advances in the epidemiology and genetics of acute intermittent porphyria. Intractable Rare Dis. Res. 2020, 9, 196–204.
- Lenglet, H.; Schmitt, C.; Grange, T.; Manceau, H.; Karboul, N.; Bouchet-Crivat, F.; Robreau, A.M.; Nicolas, G.; Lamoril, J.; Simonin, S.; et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum. Mol. Genet. 2018, 27, 1164–1173.
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2013, 36, 849–857.
- Baumann, K.; Kauppinen, R. Penetrance and predictive value of genetic screening in acute porphyria. Mol. Genet. Metab. 2020, 130, 87–99.
- Andersson, C.; Floderus, Y.; Wikberg, A.; Lithner, F. The W198X and R173W mutations in the porphobilinogen deaminase gene in acute intermittent porphyria have higher clinical penetrance than R167W. A population-based study. Scand. J. Clin. Lab. Investig. 2000, 60, 643–648.
- Fu, Y.; Jia, J.; Yue, L.; Yang, R.; Guo, Y.; Ni, X.; Shi, T. Systematically Analyzing the Pathogenic Variations for Acute Intermittent Porphyria. Front. Pharmacol. 2019, 10, 1018.
- Szlendak, U.; Bykowska, K.; Lipniacka, A. Clinical, Biochemical and Molecular Characteristics of the Main Types of Porphyria. Adv. Clin. Exp. Med. 2016, 25, 361–368.
- Brennan, M.J.; Cantrill, R.C. Delta-Aminolaevulinic acid and amino acid neurotransmitters. Mol. Cell. Biochem. 1981, 38 Pt 1, 49–58.
- Laafi, J.; Homedan, C.; Jacques, C.; Gueguen, N.; Schmitt, C.; Puy, H.; Reynier, P.; Carmen Martinez, M.; Malthiery, Y. Pro-oxidant effect of ALA is implicated in mitochondrial dysfunction of HepG2 cells. Biochimie 2014, 106, 157–166.
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019.
- Onuki, J.; Chen, Y.; Teixeira, P.C.; Schumacher, R.I.; Medeiros, M.H.; Van Houten, B.; Di Mascio, P. Mitochondrial and nuclear DNA damage induced by 5-aminolevulinic acid. Arch. Biochem. Biophys. 2004, 432, 178–187.
- Dowman, J.K.; Gunson, B.K.; Mirza, D.F.; Bramhall, S.R.; Badminton, M.N.; Newsome, P.N.; on behalf of the UK Liver Selection and Allocation Working Party. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transplant. 2012, 18, 195–200.
- Dowman, J.K.; Gunson, B.K.; Bramhall, S.; Badminton, M.N.; Newsome, P.N. Liver transplantation from donors with acute intermittent porphyria. Ann. Intern. Med. 2011, 154, 571–572.
- Andersson, C.; Bylesjö, I.; Lithner, F. Effects of diabetes mellitus on patients with acute intermittent porphyria. J. Intern. Med. 1999, 245, 193–197.
- Lin, C.-N.; Shiao, M.-S.; Cheng, M.-L.; Chen, C.-M.; Kuo, H.-C. Profiling of Serum Metabolites of Acute Intermittent Porphyria and Asymptomatic HMBS Mutation Carriers. Cells 2021, 10, 2579.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
4 times
(View History)
Update Date:
15 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No