 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marco Milanese | + 1393 word(s) | 1393 | 2021-11-15 04:27:08 | | | |
| 2 | Peter Tang | + 5 word(s) | 1398 | 2022-03-14 03:11:18 | | |
Video Upload Options
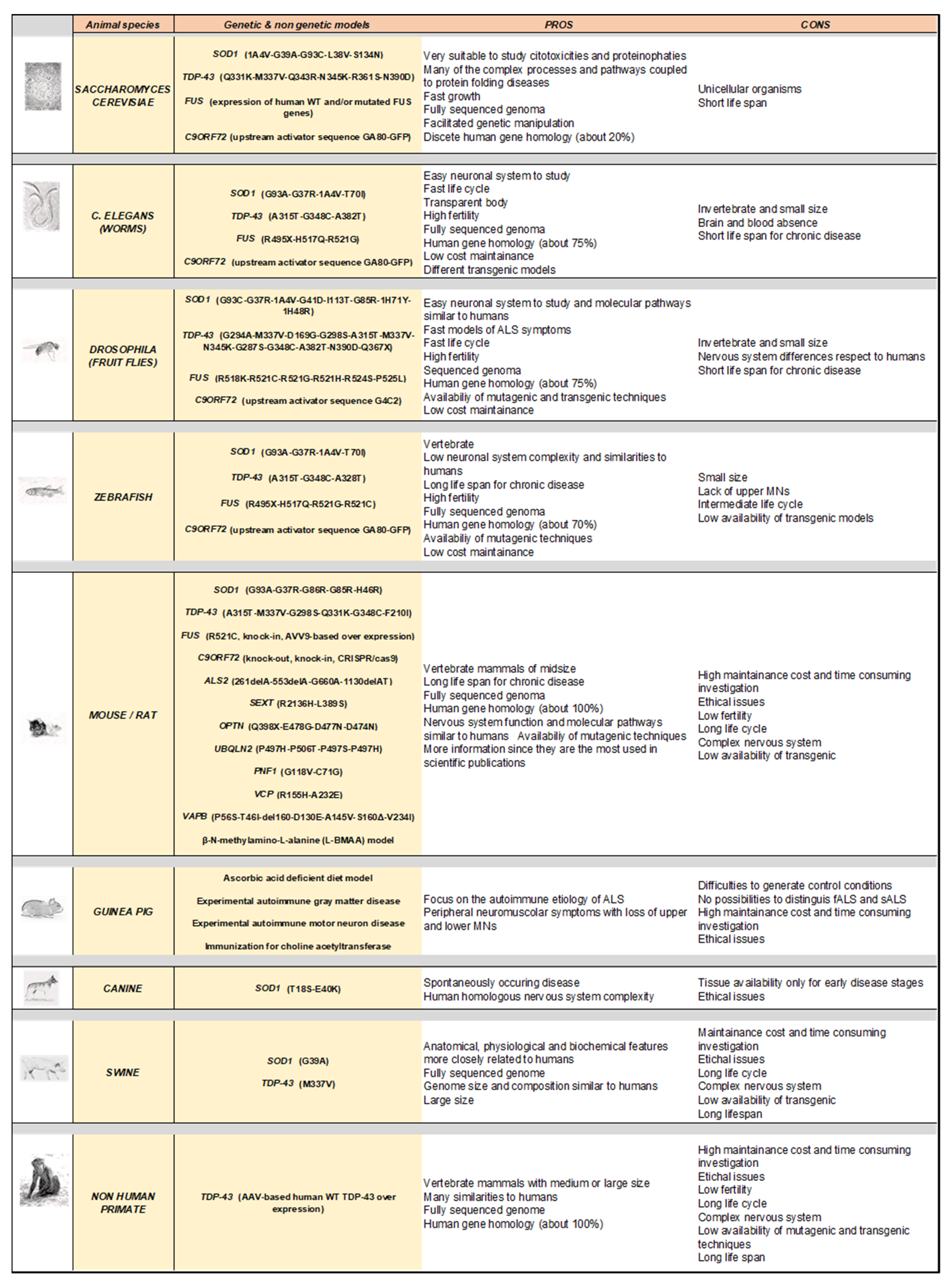
Amyotrophic lateral sclerosis (ALS) is a fatal, multigenic, multifactorial, and non-cell autonomous neurodegenerative disease characterized by upper and lower motor neuron loss. Several genetic mutations lead to ALS development and many emerging gene mutations have been discovered in recent years. Over the decades since 1990, several animal models have been generated to study ALS pathology including both vertebrates and invertebrates such as yeast, worms, flies, zebrafish, mice, rats, guinea pigs, dogs, and non-human primates. Although these models show different peculiarities, they are all useful and complementary to dissect the pathological mechanisms at the basis of motor neuron degeneration and ALS progression, thus contributing to the development of new promising therapeutics.
1. Introduction
2. Genes Involved in ALS
3. Modelling Human ALS in Living Organisms
4. Conclusions
References
- Brown, R.H.J.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602.
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17085.
- Štětkářová, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231.
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628.
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Dukkipati, S.S.; Garrett, T.L.; Elbasiouny, S.M. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 1723–1745.
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042.
- Pieri, M.; Caioli, S.; Canu, N.; Mercuri, N.B.; Guatteo, E.; Zona, C. Over-expression of N-type calcium channels in cortical neurons from a mouse model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2013, 247, 349–358.
- Bonifacino, T.; Musazzi, L.; Milanese, M.; Seguini, M.; Marte, A.; Gallia, E.; Cattaneo, L.; Onofri, F.; Popoli, M.; Bonanno, G. Altered mechanisms underlying the abnormal glutamate release in amyotrophic lateral sclerosis at a pre-symptomatic stage of the disease. Neurobiol. Dis. 2016, 95, 122–133.
- Sugiyama, K.; Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 2018, 497, 689–693.
- Tedeschi, V.; Petrozziello, T.; Secondo, A. Calcium Dyshomeostasis and Lysosomal Ca(2+) Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1216.
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90.
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis regulates trafficking of GABA and glycine heterotransporters in spinal cord glutamatergic synapses: A mechanism for the excessive heterotransporter-induced release of glutamate in experimental amyotrophic lateral sclerosis. Neurobiol. Dis. 2015, 74, 314–324.
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Milanese, M.; Bonifacino, T.; Bonanno, G.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of calpain-1 in the early phase of experimental ALS. Arch. Biochem. Biophys. 2014, 562, 1–8.
- Hadzhieva, M.; Kirches, E.; Wilisch-Neumann, A.; Pachow, D.; Wallesch, M.; Schoenfeld, P.; Paege, I.; Vielhaber, S.; Petri, S.; Keilhoff, G.; et al. Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 2013, 230, 94–101.
- Burlando, B.; Milanese, M.; Giordano, G.; Bonifacino, T.; Ravera, S.; Blanchini, F.; Bonanno, G. A multistationary loop model of ALS unveils critical molecular interactions involving mitochondria and glucose metabolism. PLoS ONE 2020, 15, e0244234.
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901.
- Morfini, G.A.; Bosco, D.A.; Brown, H.; Gatto, R.; Kaminska, A.; Song, Y.; Molla, L.; Baker, L.; Marangoni, M.N.; Berth, S.; et al. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS ONE 2013, 8, e65235.
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 2018, 9, 596.
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877.
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 2020, 14, 194.
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117.
- Chiarotto, G.B.; Cartarozzi, L.P.; Perez, M.; Biscola, N.P.; Spejo, A.B.; Gubert, F.; França Junior, M.; Mendez-Otero, R.; de Oliveira, A.L.R. Tempol improves neuroinflammation and delays motor dysfunction in a mouse model (SOD1(G93A)) of ALS. J. Neuroinflamm. 2019, 16, 218.
- Zetterström, P.; Stewart, H.G.; Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Oliveberg, M.; Marklund, S.L. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc. Natl. Acad. Sci. USA 2007, 104, 14157–14162.
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263.
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126.
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284.
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407.
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front. Cell. Neurosci. 2014, 8, 402.
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509.
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352.
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered glucose catabolism in the presynaptic and perisynaptic compartments of SOD1(G93A) mouse spinal cord and motor cortex indicates that mitochondria are the site of bioenergetic imbalance in ALS. J. Neurochem. 2019, 151, 336–350.
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182.
- Droppelmann, C.A.; Campos-Melo, D.; Ishtiaq, M.; Volkening, K.; Strong, M.J. RNA metabolism in ALS: When normal processes become pathological. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 321–336.
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 2019, 13, 601.
- Filipi, T.; Hermanova, Z.; Tureckova, J.; Vanatko, O.; Anderova, A.M. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J. Clin. Med. 2020, 9, 261.
- Crabé, R.; Aimond, F.; Gosset, P.; Scamps, F.; Raoul, C. How Degeneration of Cells Surrounding Motoneurons Contributes to Amyotrophic Lateral Sclerosis. Cells 2020, 9, 2550.
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42.
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310.
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156.
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549.
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.A.; Turner, M.R.; et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 510–518.
- Grad, L.I.; Cashman, N.R. Prion-like activity of Cu/Zn superoxide dismutase: Implications for amyotrophic lateral sclerosis. Prion 2014, 8, 33–41.
- Chen, Q.; Zhou, J.; Huang, C.; Huang, B.; Bi, F.; Zhou, H.; Xiao, B. Temporal Expression of Mutant TDP-43 Correlates with Early Amyotrophic Lateral Sclerosis Phenotype and Motor Weakness. Curr. Neurovasc. Res. 2018, 15, 3–9.
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574.
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507.
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943.
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705.
- Kwiatkowski, T.J.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208.
- Hewitt, C.; Kirby, J.; Highley, J.R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; McDermott, C.J.; Shaw, P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 455–461.
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211.
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256.
- McGoldrick, P.; Zhang, M.; van Blitterswijk, M.; Sato, C.; Moreno, D.; Xiao, S.; Zhang, A.B.; McKeever, P.M.; Weichert, A.; Schneider, R.; et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology 2018, 90, e323–e331.
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350.
- Jiang, L.; Zhang, T.; Lu, K.; Qi, S. The progress in C9orf72 research: ALS/FTD pathogenesis, functions and structure. Small GTPases 2021, 1–21.
- van Blitterswijk, M.; van Es, M.A.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, e1–e4.
- Gitler, A.D.; Tsuiji, H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016, 1647, 19–29.
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101.
- Bruijn, L.I.; Beal, M.F.; Becher, M.W.; Schulz, J.B.; Wong, P.C.; Price, D.L.; Cleveland, D.W. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide d. Proc. Natl. Acad. Sci. USA 1997, 94, 7606–7611.
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62.
- Ezzi, S.A.; Urushitani, M.; Julien, J.-P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178.
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 938–948.
- Jhanji, R.; Behl, T.; Sehgal, A.; Bungau, S. Mitochondrial dysfunction and traffic jams in amyotrophic lateral sclerosis. Mitochondrion 2021, 58, 102–110.
- Liu, Y.-J.; Tsai, P.-Y.; Chern, Y. Energy Homeostasis and Abnormal RNA Metabolism in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2017, 11, 126.
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953.
- Leblond, C.S.; Kaneb, H.M.; Dion, P.A.; Rouleau, G.A. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 91–101.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet. Neurol. 2018, 17, 94–102.
- Schram, S.; Loeb, J.A.; Song, F. Disease propagation in amyotrophic lateral sclerosis (ALS): An interplay between genetics and environment. J. Neuroinflamm. 2020, 17, 175.
- Spencer, P.S.; Lagrange, E.; Camu, W. ALS and environment: Clues from spatial clustering? Rev. Neurol. 2019, 175, 652–663.
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58.
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21.
- Lutz, C. Mouse models of ALS: Past, present and future. Brain Res. 2018, 1693, 1–10.
- Liguori, F.; Amadio, S.; Volonté, C. Where and Why Modeling Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 3977.
- Albert C. Ludolph; Caterina Bendotti; Eran Blaugrund; Bastian Hengerer; Jean‐Philippe Löffler; Joanne Martin; Vincent Meininger; Thomas Meyer; Saliha Moussaoui; Wim Robberecht; et al.Sean ScottVincenzo SilaniLeonard H. Van Den BergENMC GROUP FOR THE ESTABLISHMENT OF Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: Report on the 142nd ENMC international workshop. Amyotrophic Lateral Sclerosis 2007, 8, 217-223, 10.1080/17482960701292837.
- John C McGrath; Elspeth M McLachlan; Rolf Zeller; Transparency in Research involving Animals: The Basel Declaration and new principles for reporting research in BJP manuscripts. British Journal of Pharmacology 2015, 172, 2427-2432, 10.1111/bph.12956.

