Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raphael Souza Pinto | + 2447 word(s) | 2447 | 2022-03-02 10:04:08 | | | |
| 2 | Peter Tang | Meta information modification | 2447 | 2022-03-11 03:33:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pinto, R. Advanced Glycation End Products and Cardiovascular Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/20451 (accessed on 23 July 2026).
Pinto R. Advanced Glycation End Products and Cardiovascular Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/20451. Accessed July 23, 2026.
Pinto, Raphael. "Advanced Glycation End Products and Cardiovascular Disease" Encyclopedia, https://encyclopedia.pub/entry/20451 (accessed July 23, 2026).
Pinto, R. (2022, March 10). Advanced Glycation End Products and Cardiovascular Disease. In Encyclopedia. https://encyclopedia.pub/entry/20451
Pinto, Raphael. "Advanced Glycation End Products and Cardiovascular Disease." Encyclopedia. Web. 10 March, 2022.
Copy Citation
Epidemiological studies demonstrate the role of early and intensive glycemic control in the prevention of micro and macrovascular disease in both type 1 and type 2 diabetes mellitus (DM). Hyperglycemia elicits several pathways related to the etiopathogenesis of cardiovascular disease (CVD), including the generation of advanced glycation end products (AGEs).
advanced glycation end-products
diabetes mellitus
cardiovascular disease
atherosclerosis
reverse cholesterol transport
1. Introduction
The last two decades have been marked by alarming data about the epidemic of diabetes mellitus (DM). In 2021, an estimated 537 million adults aged 20–79 years have DM. This represents a prevalence of 10.5% of the world’s population in this age group. This number is projected to reach 643 million by 2030, and 783 million by 2045, according to the International Diabetes Federation (IDF) [1].
DM confers a higher risk of development of chronic complications and people with DM are 2 to 3 times more likely to have cardiovascular disease (CVD) [2][3]; the prevalence of end-stage renal disease is 10 times higher [4] and amputation is 10 to 20 times more common [5] in people with DM as compared to the healthy population. In addition, diabetic retinopathy is the leading cause of vision loss in working-age adults [6].
The classic studies, namely the United Kingdom Prospective Diabetes Study (UKPDS), Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE), Action to Control Cardiovascular Risk in Diabetes (ACCORD), and Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC), demonstrated that intensive glycemic therapy in patients with a recent diagnosis of type 1 and 2 diabetes (T2DM) was associated with a reduced risk in long-term complications, in particular, microvascular morbidities. Data from 30 years of the DCCT/EDIC demonstrated that intensive versus conventional glycemic treatment also has beneficial effects on macrovascular disease, especially on subclinical markers of atherosclerosis (carotid intima-media thickness and coronary calcium score) 6–12 years after the end of randomized treatment. Intensive treatment reduced aggregate cardiovascular risk by 42% and major cardiovascular events by 57% (myocardial infarction, stroke, and cardiovascular death) [7]. However, in many cases, when the glycemic control was achieved after a period of intense decompensation, the development of chronic complications was not well prevented [8][9]. Those findings seem to rely on the concept of metabolic memory or legacy effect that is based on both the permanent modification of biological macromolecules during the period of glycemic decompensation and on the accumulated risk memorized by long-term exposure to oxidative stress and advanced glycation products (AGEs), which may favor epigenetic changes. The risk of complications remains even with multifactorial interventions based on glycemic-reducing agents and antilipemic and antihypertensive drugs [8][10].
2. Formation of Advanced Glycation End Products
Louis Camille Maillard first described in 1912 the nonenzymatic reaction between reducing sugars with foods in what was called a browning reaction. Patton and Hill (1948) [11] characterized the chemical reaction that attained much more attention by the identification by Rahbar et al. (1969) [12] of a glucose-modified form of hemoglobin (glycated hemoglobin) that was found elevated in the blood of subjects with DM as compared to healthy controls. Since then, HbA1c was utilized as a parameter of metabolic control and management of DM and was associated with the development of DM complications.
The covalent and nonenzymatic reaction between reducing sugars, including glucose, with the amino-terminal portion of lysine and arginine residues in proteins, phospholipids, and nucleic acids leads to the formation of an unstable Schiff Base. The Schiff Base undergoes a more stable Amadori product or fructosyl lysine (such as glycated hemoglobin and fructosamine) and this reaction primarily depends on plasma glucose levels and the time of DM decompensation. Molecular intra and inter rearrangements of the Amadori product in most of the cases involve oxidative reactions leading to the generation of oxoaldehyde that induces a rapid and irreversible modification of proteins by advanced glycation. Oxoaldehydes are very reactive compounds, such as glyoxal (GO), methylglyoxal (MGO), glycolaldehyde, and 3-deoxyglucosone (3-DG), which are generated in DM, but also in other metabolic conditions where the carbonyl stress prevails. They rapidly interact with macromolecules leading to the fast formation of extracellular and intracellular AGEs (Figure 1).
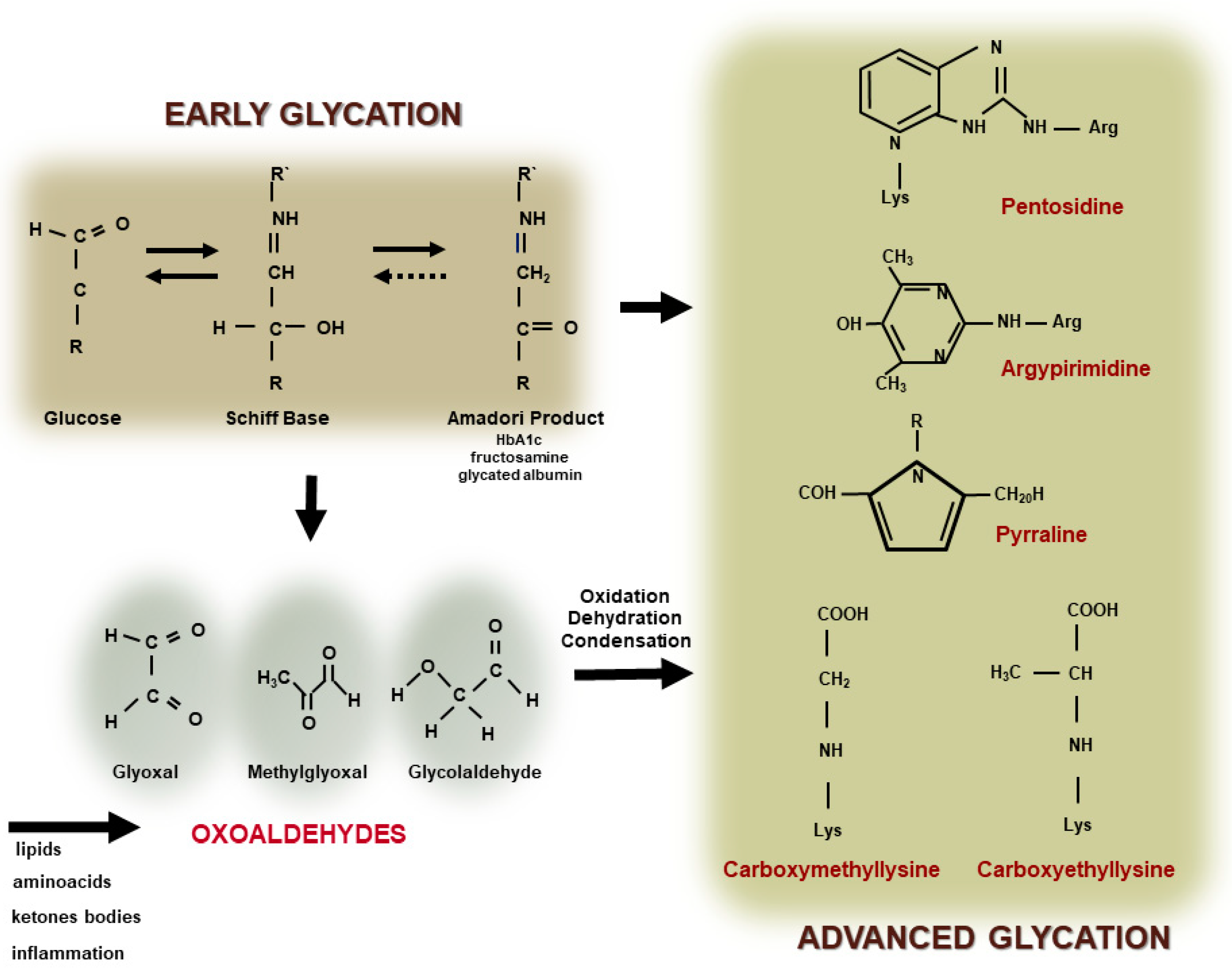
Figure 1. Advanced glycation end product (AGE) formation. The glycation reaction takes place by the modification of amino-terminal groups of proteins, phospholipids, or nucleic acids, by glucose or other monosaccharides leading to the generation of a Schiff Base and an Amadori product (early glycation). The auto-oxidation of glucose and rearrangements of the Schiff Base and the Amadori product, as well as the oxidation of amino acids, lipids or ketone bodies, and inflammation, promotes the generation of oxoaldehydes (such as glyoxal, methylglyoxal, and glycolaldehyde). They lead to the rapid and irreversible generation of AGEs, including heterogenous compounds, such as, pentosidine, argypirimidine, pyrraline, carboxymethyllysine, and carboxyethyllysine.
Intracellular generated AGEs originate from the increased glucose flow through glycolysis that favor the output of reactive oxygen species (ROS) from mitochondria and the DNA-repairing enzyme poly (ADP-ribose) polymerase (PARP). Although protecting DNA from ROS-induced damage, PARP promotes poly ribosylation of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH), impairing its activity. Substrate deviation from glycolysis generates MGO and, as a consequence, AGEs are formed feeding a vicious circle by inducing oxidative stress [13][14]. Hyperglycemia also induces the activation of the hexosamine, polyol and protein kinase C pathways, exacerbating the intracellular oxidative stress, which perpetuates the AGE generation [13][15] (Figure 2).
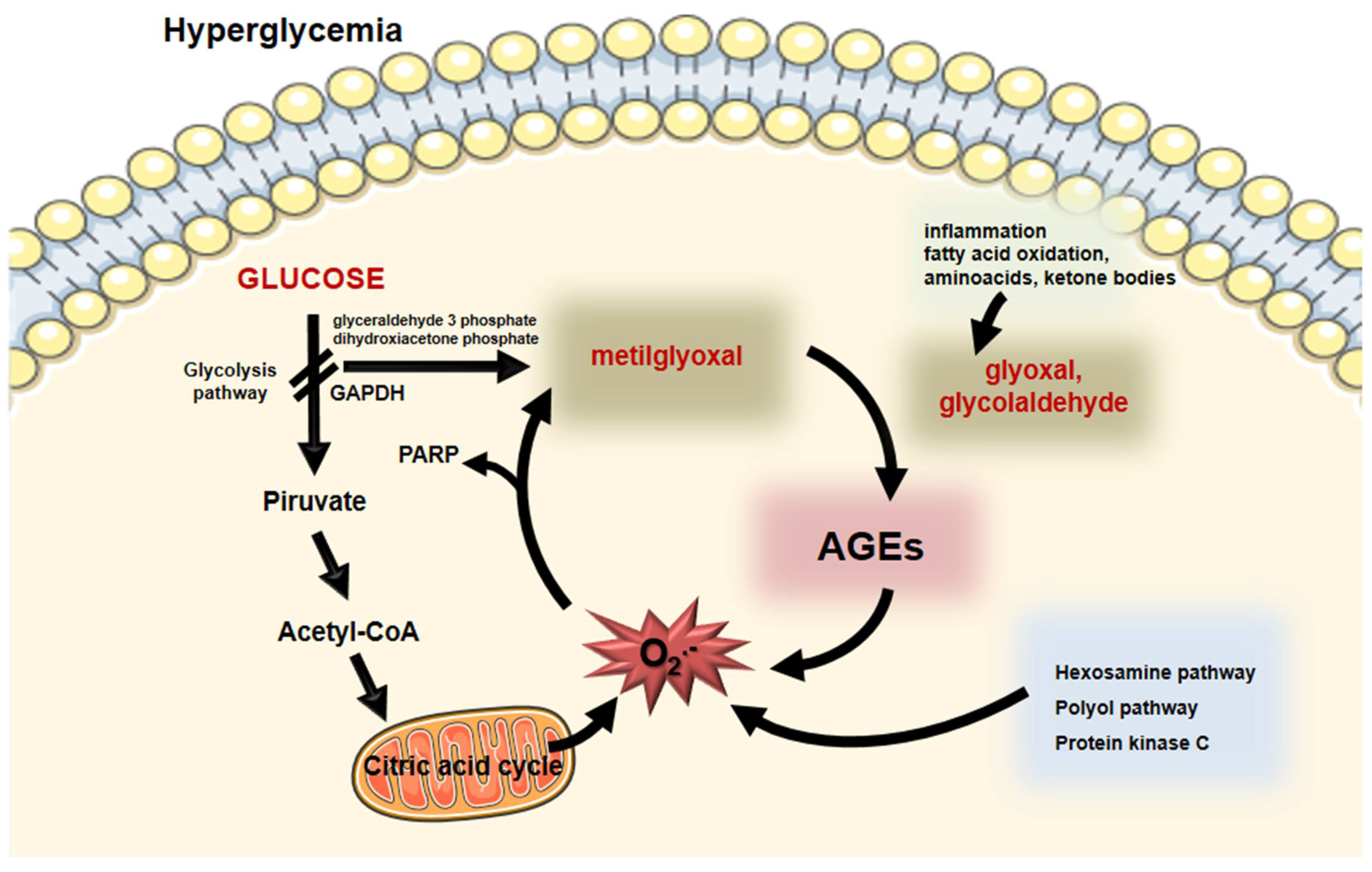
Figure 2. Intracellular formation of AGEs. During hyperglycemia, enhanced glucose flux through glycolysis induces the generation of superoxide anion by the mitochondria. Poly (ADP- ribose) polymerase (PARP) that protects DNA from cleavage induces a posttranslational modification of the glyceraldehyde 3P-dehydrogenase (GADPH) impairing glycolysis. Substrate (glyceraldehyde 3 phosphate and dihydroxyacetone phosphate) deviation leads to the generation of methylglyoxal that induces the formation of AGEs. Moreover, inflammation, fatty acid, amino acid, and ketone body oxidation generate oxoaldehydes, including glyoxal and glycolaldehyde that promote AGE generation. Oxidative stress increases by AGEs and by other biochemical pathways elicited by hyperglycemia (hexosamine, polyol, and protein kinase C). This vicious circle feeds the formation of AGEs and oxidative stress that is the base of cellular complications in diabetes mellitus. Parts of the figure were drawn using Servier Medical Art (https://smart.servier.com/, accessed on 14 December 2021).
Independently of hyperglycemia, AGEs are induced during acute and chronic inflammation due to the activation of the neutrophil myeloperoxidase that induces glycolaldehyde (GAD), another very reactive oxoaldehyde. Additionally, the oxidation of certain amino acids, polyunsaturated fatty acids, and ketone bodies contributes to the generation of oxoaldehydes and then to AGE generation. In chronic kidney disease, AGEs are formed due to a reduction in the clearance of glycation reaction precursors and reduced antioxidant enzymes. Then, although the glycation reaction was firstly ascribed to a hyperglycemic milieu, it is now conceivable that it also occurs independently of glucose concentration in plasma and tissues [16]. In fact, glycation is prevalent in healthy aging individuals, mainly in association with long half-life protein, such as collagen and crystalline. Although this process takes place continuously in the body during aging, it is extremely accelerated in DM and in other carbonyl stress conditions.
Exogenous sources of oxoaldehydes and AGEs include diet and tobacco, representing unpredictable sources of the AGEs that contribute the body’s AGE pool, although the exact mechanisms that regulate AGE absorption are not well known. The impact of AGE-containing diets on human health is under intensive investigation. High heating, dry cooking, and long-time processing increase AGEs in baked, broiled, grilled, and fried foods. Cooking in an acidic medium containing wine, vinegar, or citrus juice helps to prevent AGE formation in food. A recent publication demonstrated an endocytic pathway via scavenger receptors mediating the intestinal absorption of dietary carboxymethyllysine in C. elegans [17]. In healthy humans, about 10% of dietary AGEs are absorbed and transported in circulation in association with albumin and lipoproteins, and 30% is eliminated in the urine. On the other hand, in individuals with kidney disease, only 5% of dietary AGE is excreted in the urine [18]. A high-AGE diet results in significant elevations in serum AGE and induces oxidative stress [19]. Oxoaldehydes increase in the postprandial states and during glycemic excursions inducing a fast generation of AGEs in circulation [20]. Glucose fluctuations increase plasma levels of glyceraldehyde-derived advanced glycation end-products and relate to the severity of cardiovascular disease in DM [21].
Advanced glycation end products are very heterogeneous, making difficult their assessment in blood, tissues, and cells. Some of them induces protein crosslinking and/or emit fluorescence [22]. Glyoxal and GAD render carboxymethyl lysine (CML) the most abundant AGE found in the body [23][24], particularly in the target tissues of DM late complications, such as kidneys, eyes, skin, and vessels [25][26][27][28][29]. Methylglyoxal renders to carboxyethyl lysine, MGO-derived hydroimidazolone 1 (MGO-H1), and MGO-lysine dimer, although other structures are also described in plasma and tissues, such as pentosidine, pirralyne, argypirimidine, and GO- lysine dimer.
AGEs are recognized by several receptors, although the signaling pathway elicited is better described for the advanced glycosylation end product-specific receptor, (alias RAGE). In fact, it is demonstrated that AGER mediates the biological effects of AGEs in many cell types. AGER is a multiligand receptor of the immunoglobulin superfamily, a member of pattern-recognition receptors that recognize AGEs, but also calgranulins, high mobility group protein B1 (HMGB1), lipopolysaccharides (LPS), sheet fibrils, and phosphatidylserine on the surface of apoptotic cells. The receptor for AGEs actively participates in diabetic vascular complications, as well as in the interface of innate and adaptive immunity and in inflammation. In this sense, there is a lot of evidence supporting the concept that AGEs and AGERs play an active role in the development and progression of cardiovascular disease in DM [30].
3. AGEs as Indicators of Cardiovascular Burden: Where Are We?
The body pool of AGEs is determined by carbonyl and inflammatory stress together with kidney function that drives endogenous AGEs formation. In addition, exogenous sources that are in most cases unpredictable and variable add to this pool worsening chronic complications elicited by the AGE accumulation (Figure 3).
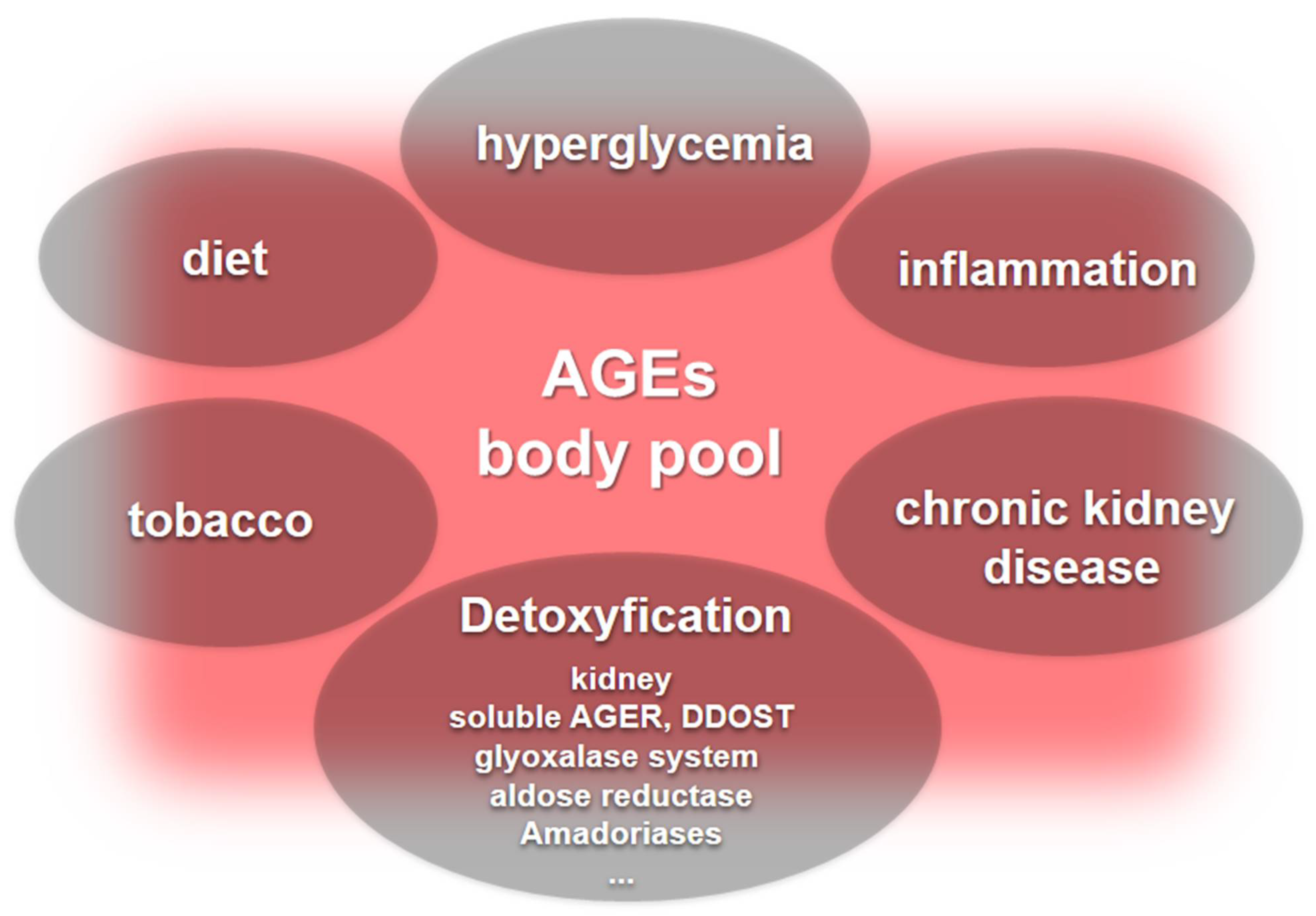
Figure 3. The body pool of AGEs. Hyperglycemia, inflammation, and chronic kidney disease are major contributors to the body’s pool of AGEs. Furthermore, dietary sources of AGEs and tobacco add to the AGE pool, while detoxification systems include kidney function and the role played by soluble AGERs and DDOST that neutralize deleterious AGER signaling, along with enzyme systems, including the glyoxalase system, aldose reductase, and the lesser-known amadoriases.
Markers of early glycation, such as HbA1c, fructosamine, and glycated albumin, were associated with vascular outcomes and mortality in the community-based Atherosclerosis Risk in Communities (ARIC) study [31]. Additionally, AGEs independently relate to atherosclerosis in both DM and non-DM subjects [32][33][34][35]. Strong evidence demonstrates that in DM patients [36] or DM patients with micro and/or macrovascular complications [37][38] and individuals with chronic kidney disease [39], circulating AGEs are elevated and associated with the progression of complications. Particularly, CML levels predict arterial stiffness [30] and the severity of vascular obstruction [40].
4. Sweet AGEs Driving the Bitter Pathophysiology of CVD
Although firstly described in association with long half-life proteins, such as crystalline and collagen, advanced glycation also occurs in long and short half-life proteins, creating a large spectrum of biological derangements. Apolipoproteins and some phospholipids that are constituents of lipoproteins are susceptible to advanced glycation compromising their biological functions. Chylomicrons and very-low-density lipoproteins (VLDL) are less susceptible to the lipoprotein lipase-mediated hydrolysis of triglycerides favoring hypertriglyceridemia. Due to the enhanced affinity of cholesteryl ester transfer protein for glycated lipoproteins and due to hypertriglyceridemia, an enhanced exchange of esterified cholesterol and triglycerides can be observed between TG-rich lipoproteins and low (LDLs) and high-density lipoproteins (HDLs). This favors the formation of small dense LDLs that are atherogenic, being more susceptible to oxidation and to enter the arterial wall compartment. Glycated LDLs are not well recognized by the LDL receptor (B-E receptor), allowing this particle to reach the arterial wall, where they are taken up by macrophage receptors (AGERs, Toll-like receptors, and scavenger receptors class A and B) leading to intracellular cholesterol accumulation and foam cell formation.
Contrary to LDLs, which have a longer half-life when glycated, glucose-modified HDLs are faster removed from the plasma. Considering the role of this lipoprotein in the prevention of atherosclerosis, there are several investigators dealing with HDL glycation and the consequences to its function.
An HDL mediates the reverse cholesterol transport a unique system that promotes excess cholesterol exportation from arterial wall macrophages to the liver, allowing its excretion in feces. Lipid poor-apoA-I or nascent HDL particles (pre-beta HDLs) interact with the phospholipid-transporting ATPase ABCA1 (ABCA-1) that is expressed in macrophages by the liver X receptor (LXR) activation by oxysterols. Free cholesterol is esterified by the phosphatidylcholine-sterol acyltransferase (alias, lecithin cholesterol acyltransferase; LCAT) being transferred to the hydrophobic HDL core that assumes a spherical format. Larger HDL particles, especially HDL2, remove more cell cholesterol by the interaction with the ATP binding cassette transporter G-1 (ABGA-1) that also exports toxic oxysterols outside macrophages. Esterified cholesterol is transferred to apo B-containing lipoproteins (chylomicrons, VLDL and LDL) by the action of CETP; allowing for the uptake of these lipoproteins by the hepatic receptors B-E and the prolow-density lipoprotein receptor-related protein 1 (LRP-1). The scavenger receptor class B member 1 (SR-B1) in the liver selectively removes esterified cholesterol in HDLs. Cholesterol can be eliminated in bile in its free form or after conversion into cholic and deoxycholic acids, by the action of, respectively, oxysterol 7-alpha-hydroxylase and sterol 27-hydroxylase, and then excreted in feces.
Early and advanced glycation compromises many steps of the RCT contributing to atherogenesis in DM. The LCAT activity on glycated HDLs is reduced, while the CETP activity is enhanced favoring the accumulation of cholesterol in atherogenic lipoproteins (LDLs, VLDLs, and chylomicrons) [41]. In addition, advanced glycated HDLs remove less cholesterol from macrophages, and the esterified cholesterol delivery to the liver mediated by SRB1 is reduced [42] (Figure 4).
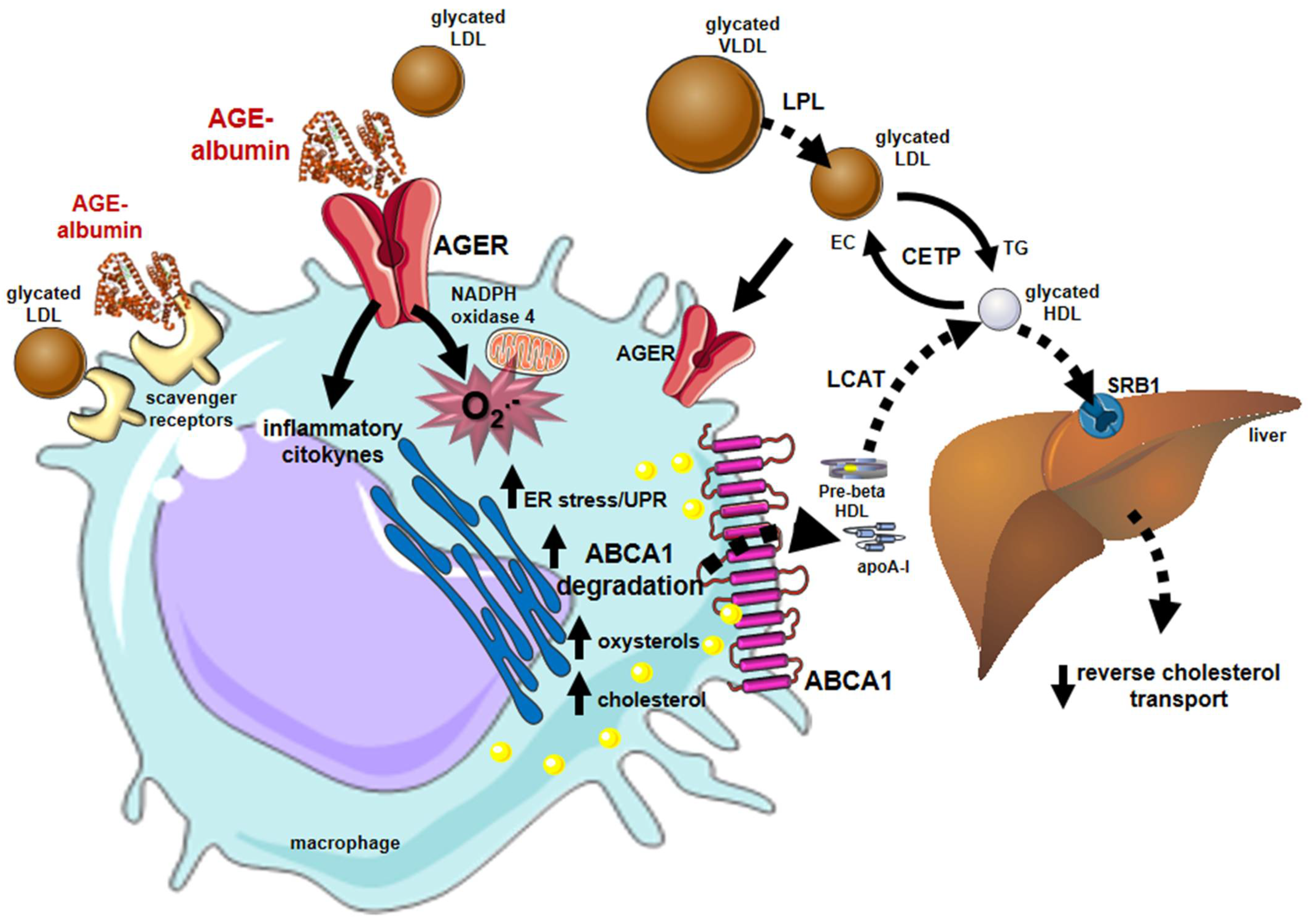
Figure 4. Major influences of glycation on lipid metabolism and reverse cholesterol transport that favor atherogenesis. Alterations in lipid and lipoprotein metabolism induced by early and advanced glycation favor atherogenesis. In blood circulation, the lipolysis of chylomicrons and VLDLs by the lipoprotein lipase (LPL) is damaged by glycation, inducing plasma triglycerides elevation. The activity of CETP is also stimulated between glycated lipoproteins enabling more cholesterol to be transferred from HDLs to atherogenic lipoproteins and reducing HDL cholesterol. Glycated LDLs are taken up by macrophage scavenger receptors and by the advanced glycosylation end product-specific receptor (AGER). Advanced glycated albumin (AGE-albumin) induces, via AGERs, oxidative stress by enhancing the activity of NADPH oxidase 4 and mitochondrial systems and inflammation. Together, oxidative stress and inflammation lead to ER stress that triggers proteasomal and lysosomal degradation of the ABCA-1 compromising the cholesterol efflux to apo A-I and the generation of HDLs. The activity of (LCAT) is impaired by HDL glycation reducing the lipoprotein maturation. The accumulation of cholesterol and toxic oxysterols in macrophages triggers oxidation, inflammation, and ER stress creating a vicious circle that contributes to atherogenesis, plaque instability, and rupture. Parts of the figure were drawn using Servier Medical Art (https://smart.servier.com/, accessed on 14 December 2021).
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2021, 183, 109119.
- Danaei, G.; Lawes, C.M.M.; Vander Hoorn, S.; Murray, C.J.L.; Ezzati, M. Global and Regional Mortality from Ischaemic Heart Disease and Stroke Attributable to Higher-than-Optimum Blood Glucose Concentration: Comparative Risk Assessment. Lancet 2006, 368, 1651–1659.
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.K.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes Mellitus, Fasting Blood Glucose Concentration, and Risk of Vascular Disease: A Collaborative Meta-Analysis of 102 Prospective Studies. Lancet 2010, 375, 2215–2222.
- Collins, A.J.; Foley, R.N.; Gilbertson, D.T.; Chen, S.-C. United States Renal Data System Public Health Surveillance of Chronic Kidney Disease and End-Stage Renal Disease. Kidney Int. Suppl. 2015, 5, 2–7.
- Moxey, P.W.; Gogalniceanu, P.; Hinchliffe, R.J.; Loftus, I.M.; Jones, K.J.; Thompson, M.M.; Holt, P.J. Lower Extremity Amputations—A Review of Global Variability in Incidence. Diabet. Med. 2011, 28, 1144–1153.
- Congdon, N.G.; Friedman, D.S.; Lietman, T. Important Causes of Visual Impairment in the World Today. JAMA 2003, 290, 2057–2060.
- Lachin, J.M.; Orchard, T.J.; Nathan, D.M.; DCCT/EDIC Research Group. Update on Cardiovascular Outcomes at 30 Years of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study. Diabetes Care 2014, 37, 39–43.
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653.
- Gaede, P.; Lund-Andersen, H.; Parving, H.-H.; Pedersen, O. Effect of a Multifactorial Intervention on Mortality in Type 2 Diabetes. N. Engl. J. Med. 2008, 358, 580–591.
- Ceriello, A.; Gallo, M.; Armentano, V.; Perriello, G.; Gentile, S.; De Micheli, A.; Associazione Medici Diabetologi. Personalizing Treatment in Type 2 Diabetes: A Self-Monitoring of Blood Glucose Inclusive Innovative Approach. Diabetes Technol. Ther. 2012, 14, 373–378.
- Patton, A.R.; Hill, E.G. Inactivation of Nutrients by Heating With Glucose. Science 1948, 107, 68–69.
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an Unusual Hemoglobin in Patients with Diabetes Mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843.
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820.
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461.
- Mora-Fernández, C.; Domínguez-Pimentel, V.; de Fuentes, M.M.; Górriz, J.L.; Martínez-Castelao, A.; Navarro-González, J.F. Diabetic Kidney Disease: From Physiology to Therapeutics. J. Physiol. 2014, 592, 3997–4012.
- Thornalley, P.J.; Rabbani, N. Progress in Uremic Toxin Research: Highlights and Hotspots of Protein Glycation in End-Stage Renal Disease. Semin. Dial. 2009, 22, 400–404.
- Dubois, C.; Litke, R.; Rianha, S.; Paul-Constant, C.; Lo Guidice, J.M.; Taront, S.; Tessier, F.J.; Boulanger, E.; Fradin, C. Exposure of Caenorhabditis elegans to Dietary Nε-Carboxymethyllysine Emphasizes Endocytosis as a New Route for Intestinal Absorption of Advanced Glycation End Products. Nutrients 2021, 13, 4398.
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479.
- Cai, W.; Gao, Q.D.; Zhu, L.; Peppa, M.; He, C.; Vlassara, H. Oxidative stress-inducing carbonyl compounds from common foods: Novel mediators of cellular dysfunction. Mol. Med. 2002, 8, 337–346.
- Maessen, D.E.; Hanssen, N.M.; Scheijen, J.L.; van der Kallen, C.J.; van Greevenbroek, M.M.; Stehouwer, C.D.; Schalkwijk, C.G. Post-Glucose Load Plasma α-Dicarbonyl Concentrations Are Increased in Individuals With Impaired Glucose Metabolism and Type 2 Diabetes: The CODAM Study. Diabetes Care 2015, 38, 913–920.
- Watanabe, M.; Kawai, Y.; Kitayama, M.; Akao, H.; Motoyama, A.; Wakasa, M.; Saito, R.; Aoki, H.; Fujibayashi, K.; Tsuchiya, T.; et al. Diurnal glycemic fluctuation is associated with severity of coronary artery disease in prediabetic patients: Possible role of nitrotyrosine and glyceraldehyde-derived advanced glycation end products. J. Cardiol. 2017, 69, 625–631.
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; DCCT/EDIC Research Group; Monnier, V.M. Skin Advanced Glycation End Products Glucosepane and Methylglyoxal Hydroimidazolone Are Independently Associated with Long-Term Microvascular Complication Progression of Type 1 Diabetes. Diabetes 2015, 64, 266–278.
- Ahmed, M.U.; Thorpe, S.R.; Baynes, J.W. Identification of N Epsilon-Carboxymethyllysine as a Degradation Product of Fructoselysine in Glycated Protein. J. Biol. Chem. 1986, 261, 4889–4894.
- Vlassara, H.; Palace, M.R. Diabetes and Advanced Glycation Endproducts. J. Intern. Med. 2002, 251, 87–101.
- Baidoshvili, A.; Niessen, H.W.M.; Stooker, W.; Huybregts, R.A.J.M.; Hack, C.E.; Rauwerda, J.A.; Meijer, C.J.L.M.; Eijsman, L.; van Hinsbergh, V.W.M.; Schalkwijk, C.G. N(Omega)-(Carboxymethyl)Lysine Depositions in Human Aortic Heart Valves: Similarities with Atherosclerotic Blood Vessels. Atherosclerosis 2004, 174, 287–292.
- Nerlich, A.G.; Schleicher, E.D. N(Epsilon)-(Carboxymethyl)Lysine in Atherosclerotic Vascular Lesions as a Marker for Local Oxidative Stress. Atherosclerosis 1999, 144, 41–47.
- Thomas, C.J.; Cleland, T.P.; Sroga, G.E.; Vashishth, D. Accumulation of Carboxymethyl-Lysine (CML) in Human Cortical Bone. Bone 2018, 110, 128–133.
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2020, 10, 18.
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222.
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The Role of Advanced Glycation End-Products in the Development of Coronary Artery Disease in Patients with and without Diabetes Mellitus: A Review. Mol. Med. 2018, 24, 59.
- Selvin, E.; Rawlings, A.M.; Lutsey, P.L.; Maruthur, N.; Pankow, J.S.; Steffes, M.; Coresh, J. Fructosamine and Glycated Albumin and the Risk of Cardiovascular Outcomes and Death. Circulation 2015, 132, 269–277.
- Blaauw, J.; Smit, A.J.; van Pampus, M.G.; van Doormaal, J.J.; Aarnoudse, J.G.; Rakhorst, G.; Graaff, R. Skin Autofluorescence, a Marker of Advanced Glycation End Products and Oxidative Stress, Is Increased in Recently Preeclamptic Women. Am. J. Obstet. Gynecol. 2006, 195, 717–722.
- Chiang, K.-H.; Huang, P.-H.; Huang, S.-S.; Wu, T.-C.; Chen, J.-W.; Lin, S.-J. Plasma Levels of Soluble Receptor for Advanced Glycation End Products Are Associated with Endothelial Function and Predict Cardiovascular Events in Nondiabetic Patients. Coron. Artery Dis. 2009, 20, 267–273.
- Baumann, M.; Richart, T.; Sollinger, D.; Pelisek, J.; Roos, M.; Kouznetsova, T.; Eckstein, H.-H.; Heemann, U.; Staessen, J.A. Association between Carotid Diameter and the Advanced Glycation End Product N-Epsilon-Carboxymethyllysine (CML). Cardiovasc. Diabetol. 2009, 8, 45.
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma Carboxymethyl-Lysine, an Advanced Glycation End Product, and All-Cause and Cardiovascular Disease Mortality in Older Community-Dwelling Adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880.
- Odani, H.; Iijima, K.; Nakata, M.; Miyata, S.; Kusunoki, H.; Yasuda, Y.; Hiki, Y.; Irie, S.; Maeda, K.; Fujimoto, D. Identification of N(Omega)-Carboxymethylarginine, a New Advanced Glycation Endproduct in Serum Proteins of Diabetic Patients: Possibility of a New Marker of Aging and Diabetes. Biochem. Biophys. Res. Commun. 2001, 285, 1232–1236.
- Sampathkumar, R.; Balasubramanyam, M.; Rema, M.; Premanand, C.; Mohan, V. A Novel Advanced Glycation Index and Its Association with Diabetes and Microangiopathy. Metabolism 2005, 54, 1002–1007.
- Guerin-Dubourg, A.; Cournot, M.; Planesse, C.; Debussche, X.; Meilhac, O.; Rondeau, P.; Bourdon, E. Association between Fluorescent Advanced Glycation End-Products and Vascular Complications in Type 2 Diabetic Patients. Biomed. Res. Int. 2017, 2017, 7989180.
- Kratochvilová, M.; Zakiyanov, O.; Kalousová, M.; Kříha, V.; Zima, T.; Tesař, V. Associations of Serum Levels of Advanced Glycation End Products with Nutrition Markers and Anemia in Patients with Chronic Kidney Disease. Ren Fail. 2011, 33, 131–137.
- Kiuchi, K.; Nejima, J.; Takano, T.; Ohta, M.; Hashimoto, H. Increased Serum Concentrations of Advanced Glycation End Products: A Marker of Coronary Artery Disease Activity in Type 2 Diabetic Patients. Heart 2001, 85, 87–91.
- Passarelli, M.; Catanozi, S.; Nakandakare, E.R.; Rocha, J.C.; Morton, R.E.; Shimabukuro, A.F.; Quintão, E.C. Plasma Lipoproteins from Patients with Poorly Controlled Diabetes Mellitus and “in Vitro” Glycation of Lipoproteins Enhance the Transfer Rate of Cholesteryl Ester from HDL to Apo-B-Containing Lipoproteins. Diabetologia 1997, 40, 1085–1093.
- Matsuki, K.; Tamasawa, N.; Yamashita, M.; Tanabe, J.; Murakami, H.; Matsui, J.; Imaizumi, T.; Satoh, K.; Suda, T. Metformin Restores Impaired HDL-Mediated Cholesterol Efflux Due to Glycation. Atherosclerosis 2009, 206, 434–438.
More
Information
Subjects:
Biochemistry & Molecular Biology; Endocrinology & Metabolism; Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.3K
Revisions:
2 times
(View History)
Update Date:
11 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No