Hairy cell leukaemia is a rare chronic lymphoid malignancy with distinctive clinical and laboratory features which include an enlarged spleen, low blood counts, and infiltration of the spleen and bone marrow, with lymphocytes that have a villous or hairy cytoplasmic border. Historically it has been responsive to a range of treatment modalities including splenectomy, alpha interferon, and more recently chemotherapy, but none are curative. The Genomics of hairy cell leukaemia involves the chromosome abnormalities, genomic mutations, DNA methylation patterns, and immunoglobulin gene usage in this disease.
1. Introduction
The 2017 WHO classification of haematological malignancies recognises classical hairy cell leukaemia (HCLc) as a discrete entity and its variant form (HCLv) and splenic diffuse red pulp lymphoma (SDRPL) as provisional entities
[1]. HCLc is a rare chronic lymphoproliferative disorder, with an incidence of 0.4/100,000. It is approximately four times more common in men than women and typically presents in middle age, with fatigue, infections, and abdominal discomfort due to splenomegaly. A complete blood count frequently shows cytopenia with almost universal monocytopenia, and a blood film usually reveals small numbers of medium-sized lymphoid cells with a ‘kidney-shaped nucleus’, infrequent nucleoli, weakly basophilic cytoplasm, and hairy projections. A modest lymphocytosis is seen in 5–10% of cases. Splenic histology shows diffuse infiltration of the red pulp with atrophy of the white pulp, a pattern also seen in HCLv and SDRPL, in contrast to the predominant white pulp involvement found in Splenic Marginal Zone Lymphoma (SMZL). Although originally believed to represent a tumour of haemopoietic progenitor reticuloendothelial cells
[2], immunophenotyping identified HCLc as a tumour of mature B cells, typically expressing CD19, CD20, CD200, Tbet, PD1, and four markers of diagnostic value: CD11c and CD103, components of integrin receptors, and CD25 and CD123, components of interleukin receptors, of which at least three are expressed in all cases. Hairy cells do not express CD5 or CD27. Immunohistochemistry of bone marrow trephines additionally shows the expression of cyclin D1, CD72 (DBA-44), and Annexin A1, a specific marker for HCLc among B-cell malignancies, while bone marrow aspiration is usually unsuccessful due to the presence of reticulin fibrosis
[3][4].
HCLv has an incidence of 0.04/100,000, a median age at presentation of 70 years, and a male-to-female ratio of 1.5–2. It was first described in 1980 in two patients with bulky splenomegaly, a marked leucocytosis with villous lymphocytes, and splenic histology showing red pulp involvement similar to that seen in HCLc, together with a number of distinctive features not found in HCLc. These include the absence of monocytopenia, larger tumour cells with prominent nucleoli, and bone marrow that is easy to aspirate due to absence or minimal marrow reticulin. Immunophenotyping shows the expression of CD19, CD20, and of the four archetypal HCLc markers, only CD11c and CD103 are commonly expressed, while CD25, CD123, and CD200 are negative or only weakly expressed. Immunohistochemistry shows the expression of CD72 but not annexin A1, and cyclin D1 is negative or weak
[5][6].
In 2008, the term SDRPL was introduced to describe a further type of splenic lymphoma with circulating villous lymphocytes
[7]. The frequency of SDRPL has not been established in the general population but represented 9% of splenic B-cell lymphomas seen in a 12-year period reviewed at the Spanish National Cancer Research Centre
[8]. The clinical and laboratory features overlap those seen in HCLv, but the tumour cells generally lack a prominent nucleolus, and patients pursue a more indolent clinical course. Key demographic, morphological, phenotypic, and clinical differences between the three disorders are shown in
Table 1.
Table 1. Key Differences between HCLc, HCLv, and SDRPL.
| |
|
HCLc |
HCLv |
SDRPL |
| Demographics |
Incidence |
0.4/100,000 |
0.03/100,000 |
? |
| Median age |
55–63 |
70 |
70 |
| M:F ratio |
3–4: 1 |
1–2: 1 |
1.6–2.4: 1 |
| Haematology |
Monocytopenia |
Yes |
No |
No |
| Nucleolus |
Inconspicuous |
Single prominent |
Inconspicuous |
| Immunophenotype |
Surface IgH |
Usually, multiple isotypes |
Usually IgG +/− other isotypes |
M+/−D, M+G, G |
| CD11c |
Strong |
Strong |
Moderate |
| CD25 |
Strong |
Negative |
Negative (weak in 3%) |
| CD103 |
Strong |
Moderate |
Negative (weak in 33%) |
| CD123 |
Strong |
Negative |
Negative (weak in 15%) |
| CD27 |
Negative |
? |
Negative (positive in 20%) |
| CD200 |
Strong |
Weak or negative |
Weak |
| Immunohistochemistry |
Annexin A1 |
Positive |
Negative |
Negative |
| |
Cyclin D1 |
Positive |
Negative |
Negative |
| Outcome |
Need for treatment |
Yes |
Yes |
Approx. 50% |
2. Hairy Cell Leukaemia
2.1. BRAF V600E Mutations
The whole-exome sequencing of a single case of HCLc led to the discovery of a single somatic, point mutation in the DNA sequence of v-Raf murine sarcoma viral oncogene homolog B (
BRAF), a kinase-encoding proto-oncogene. The same mutation was subsequently found in all 47 additional cases studied. The mutation replaces thymine (T) with adenine (A) in exon 15 of
BRAF at position 1799 of the gene-coding sequence located in chromosome 7q34. In turn, this produces an amino acid change from valine (V) to glutamate (E) at position 600 (V600E) of the protein sequence, ultimately leading to aberrant activation of the
BRAF oncogenic kinase and, thus, of the downstream MEK–ERK signalling pathway, such that ERK phosphorylation (pERK), detectable by immunohistochemistry, is a ubiquitous finding in
BRAF-V600E positive HCLc
[9][10]. The
BRAF-V600E mutation in HCL is clonal and heterozygous, except in a minority of patients who lose the wild-type allele as a result of a concomitant 7q deletion
[11]. Details of the RAS–RAF–MEK–ERK pathway and the
BRAF protein with the site of the
BRAFV600E mutation are shown in
Figure 1 and
Figure 2, respectively, and described in the accompanying legends.
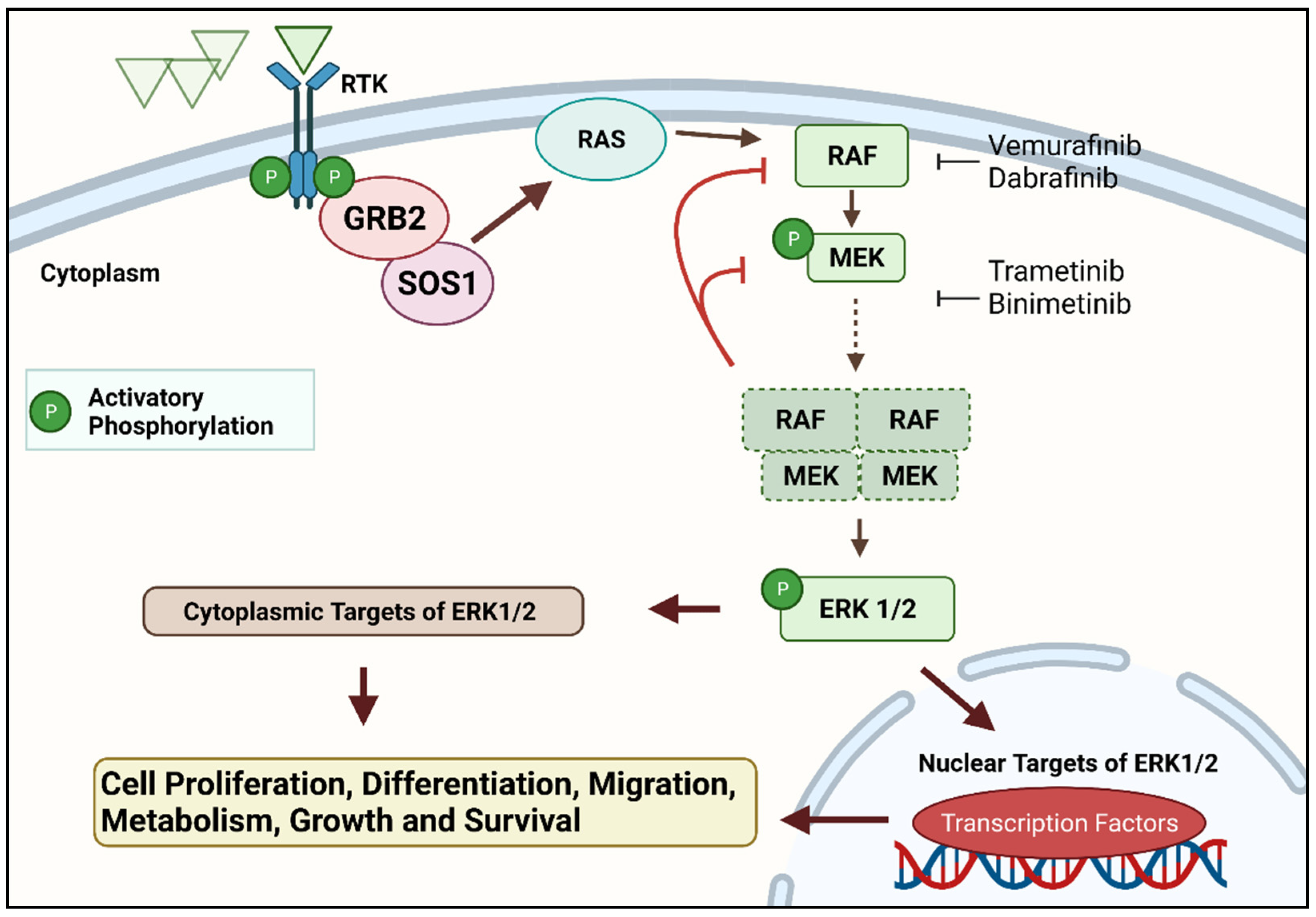
Figure 1. The RAS–RAF–MEK–ERK signal transduction cascade is one of four mitogen-activated protein kinase (MAPK) cascades which are activated in response to extracellular signals. RAS activation occurs within biomolecular condensates at the inner part of the cell membrane and recruits members of the RAF kinase family (A-RAF, B-RAF, and C-RAF/RAF-1) to the plasma membrane for activation. Active RAF kinases phosphorylate downstream mitogen-activated protein kinase/extracellular signal-regulated kinase ERK kinase (MEK). A transient tetramer, consisting of two RAF-MEK dimers, is formed to facilitate MEK activation by RAF. Active MEK then dually phosphorylates its only downstream targets, extracellular signal-related kinases 1 and 2 (ERK1/2). In contrast, ERK1/2 has extremely broad substrate specificity and is capable of activating both nuclear and cytosolic targets, many of which are transcription factors essential for the regulation of cell proliferation, survival, growth, metabolism, migration, and differentiation. In addition, ERKs also phosphorylate RAFs themselves at specific inhibitory amino acid residues, which releases RAF from RAS and extinguishes the signal via a negative feedback mechanism
[12][13]. Created with
Biorender.com (accessed on 28 December 2021).
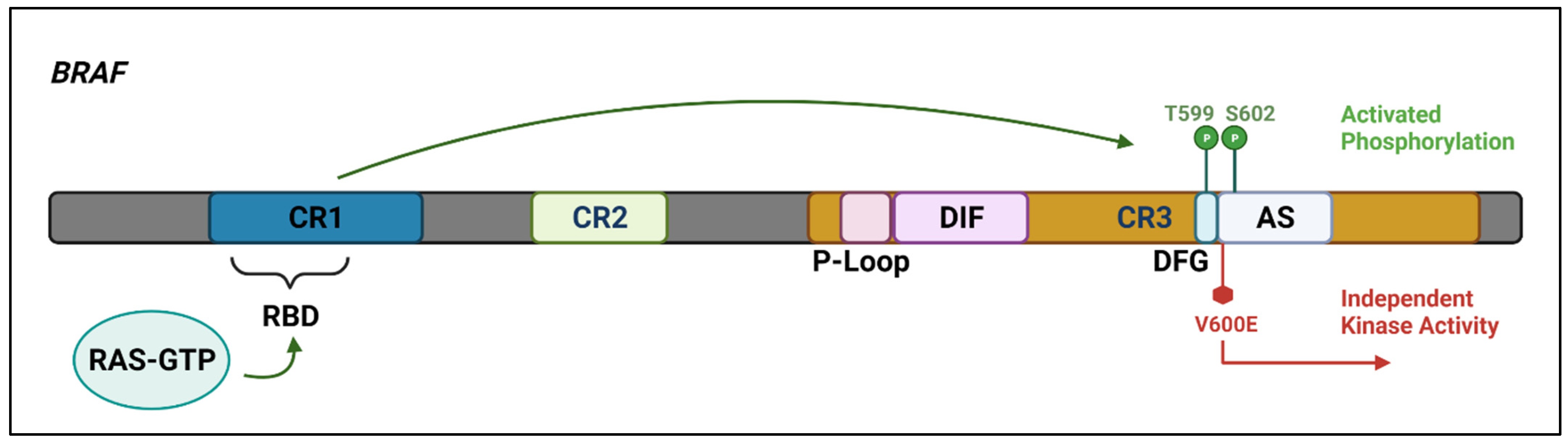
Figure 2. The
BRAF protein includes three highly conserved regions—CR1 which functions as an auto-inhibitor of the
BRAF kinase domain and contains a RAS-GTP binding domain (RBD), CR2 which acts as a flexible hinge between CR1 and CR3, and CR3, the kinase domain, which comprises multiple subregions including the P loop, the dimerisation interface (DIF), the DFG motif, and the activation segment. In the wild-type protein, inactive RAF exists in an auto-inhibited state. Under activating conditions, RAS-GTP binds to the RBD, disrupting auto-inhibition.
BRAF is then phosphorylated at T599 and S602 within the DFG motif and activation segments, destabilising interactions with the P loop and allowing the activation segment to flip into its active conformation. The majority of
BRAF mutants are located within either the P loop or the activation segment and adjacent DFG motif. The
BRAF-V600E mutation occurs in the kinase activation segment, thereby inducing a change to the active conformation independently from upstream RAS activation. This results in constitutive kinase activity and aberrant signalling through the RAF–MEK–ERK pathway
[14][15]. Created with
Biorender.com (accessed on 28 December 2021).
BRAF mutations are found in a wide range of both solid and haematopoietic tumours, with a particularly high incidence in benign melanocytic nevi, malignant melanoma, papillary carcinoma of the thyroid
[16], and the primary histiocytic disorders, Langerhans cell histiocytosis (LCH) and Erdheim–Chester disease (ECD)
[17][18]. The clinical and biological consequences of
BRAF mutations are highly variable and include the induction of a senescent phenotype, oncogenic transformation, and the emergence of secondary histiocytic sarcomas in a variety of acute or chronic, B or T cell, leukaemia, or lymphomas
[19][20][21]. This variability may reflect the acquisition of additional genomic abnormalities such as the inactivation of cell cycle inhibitors, and the differentiation stage, transcriptomic and epigenetic features of the cell type in which the
BRAF mutation arises
[22][23].
2.1.1. Haematopoietic Stem Cell Origin of BRAFV600E Mutation in HCLc
To identify the cell population from which the
BRAFV600E mutation arises, immunophenotypically distinct CD34+, CD38− lineage-negative cells which encompass haemopoietic stem cells (HSCs) and their immediate multipotent progenitors
[24][25], CD34+, CD38+ pro-B cells, myeloid progenitor cells, and HCLc cells were isolated with >97% purity from the bone marrow of 14 HCLc patients and age-matched controls
[26].
HCLc patients were characterised by an expansion of HSCs and a marked decrease in the frequency of granulocyte-macrophage progenitor cells, consistent with the neutropenia and monocytopenia characteristics of HCLc. The BRAFV600E mutation was identified in the HSC, pro-B cell, and HCL cell populations, and quantitative sequencing analysis revealed a mean BRAFV600E-mutant allele frequency of 4.97% in the HSCs. Furthermore, in one patient who also had chronic lymphocytic leukaemia (CLL), the BRAFV600E mutation was present in both tumour cell populations, consistent with the mutation arising in a common precursor. To identify additional co-occurring genetic lesions that might cooperate with the BRAFV600E mutation to promote haematopoietic transformation, targeted mutational analysis was performed on HCL cells from three patients in whom the BRAFV600E mutation had been detected in HSCs. An additional ARID1A or KMT2C mutation was present in the leukemic cells but not the HSCs of 2/3 cases. HCLc patients treated with vemurafenib showed restoration of normal myelopoiesis, demonstrating that the impaired myeloid differentiation in HCL is dependent on mutant BRAFV600E signalling. This raises the question as to whether the clinical response to BRAF inhibitors may be mediated through their effects on mature leukemic cells, as well as through targeted inhibition of signalling and survival in mutant HSPCs.
Although both arise from HSCs, the co-existence of HCLc and LCH in the same patient has rarely been reported
[27], possibly reflecting the different skewing of
BRAFV600E mutant HPC differentiation in mouse models along lymphoid or myeloid pathways in HCLc and LCH, respectively.
[26][28][29].
2.1.2. Biological Consequences of the BRAFV600E Mutation in HCLc
The biology of HCLc reflects both cell-intrinsic factors as well as interactions with antigen(s), the extracellular matrix, the multiple cell types, and their secreted products present in the tissue microenvironment (TME)
[30][31]. To ascertain the contribution of mutant
BRAF to the HCLc phenotype, hairy cells from 26 patients were exposed in vitro to the specific
BRAF inhibitors, vemurafenib or dabrafenib, or the MEK inhibitor trametinib. This resulted in the silencing of a gene expression signature which is specific to HCLc among B-cell tumours, with downregulation of genes including
CCND1, CD25, and feedback inhibitors of ERK signalling such as members of the dual-specificity phosphatase (DSP) gene family. Additionally,
BRAF or MEK inhibition caused loss of the hairy morphology and induced apoptosis which could be partially abrogated by co-culture with a bone marrow stromal cell line.
BRAF and MEK inhibitors did not elicit any of the above-described biological effects in leukemic cells from four cases with HCLv, although the
MAP2K1 mutation status of these cases was not documented
[32][33][34].
2.2. Other Genomic Abnormalities
The most frequent cytogenetic and genomic abnormalities and immunogenetic features found in BRAFV600E mutated HCLc, BRAF WT HCLc, HCLv, and SDRPL are summarised in Table 2.
Table 2. Most frequent features of HCLc, HCLv, and SDRPL.
| |
HCLc |
HCLv |
SDRPL |
HCLc BRAF
WT |
HCLc BRAF
Mutated or Upregulated |
| Recurring CNAs * |
7q Loss |
|
Y |
Y |
Y |
8q Loss
(MAPK15) |
|
Y |
N |
N |
| 17p Loss |
|
Rare |
Y |
Rare |
X or Xp Loss
(BCOR) |
|
N |
N |
Y |
| 5q Gain |
|
Y |
Y |
N |
Genomic
Mutations |
MAPK Pathway |
BRAFV600E |
0% |
>99% |
0% |
0% |
| MAP2K1 |
22% |
0% |
22–41% |
7–13% |
| Cell Cycle |
CDKN1B |
|
16% |
|
|
| CCND3 |
|
0% |
13% |
21–24% |
| Epigenetic Regulators ** |
KMT2C |
|
15% |
25% |
|
| KDM6A |
|
0% |
50% |
|
| CREBBP |
|
5% |
12–25% |
|
| ARID1A |
|
4% |
4% |
9% |
| Transcriptional Repressors |
BCOR |
|
0% |
0% |
16% |
| NFKβ Pathway |
KLF2 |
|
13% |
0% |
2% |
| Spliceosome |
U2AF1 |
|
0% |
2/7 |
0% |
| TP53 |
|
2% |
|
|
| IGHV Genes |
Homology |
100% |
|
<5% |
18–22% |
11% |
| 98–100% |
|
17–20% |
27–53% |
14% |
| <98% |
|
80% |
47–73% |
86% |
| Gene Usage |
IGHV3-23 |
|
7–10% |
|
14% |
| IGHV3-30 |
|
7–10% |
|
6% |
| IGHV4-34 |
50% |
7–10% |
17–36% |
22% |
2.3. Germline Variants
Familial HCLc exhibits similar clinical features to sporadic HCLc but is rare, with fewer than 20 families reported. Four multiplex HCLc pedigrees were recently screened for shared germline variants, conferring HCLc susceptibility. Although there was only limited overlap between the pedigrees on a variant or gene level, several functional pathways such as neutrophil-mediated immunity and G-protein-coupled receptor signalling were shared in 3/4 and MAPK and RAS signalling in 2/4 pedigrees, respectively
[35].
2.4. DNA Methylation Profile
DNA methylation profiling was analysed with the low-resolution Infinium Human Methylation27 array in 11 cases of
BRAFV600E mutated HCLc, together with cases of CLL, SMZL, and normal B-cell subsets. HCLc had a distinct global methylation profile which, nevertheless, was more closely related to SMZL than to CLL and to normal post germinal centre (GC) memory B cells and marginal zone B cells than to pre-GC and GC B cells. An integrated analysis of the HCL methylation profile and the previously published gene expression profile showed an inverse correlation between gene expression and methylation, alluding to a role for DNA promoter methylation in the regulation of specific gene expression. Gene expression patterns were consistent with constitutive activation of the RAS–RAF–MEK–ERK pathway and also affected pathways involved in the homing, migration, and survival of HCL cells
[36].
2.5. Immunogenetic Features
The analysis of immunoglobulin gene repertoires in B-cell malignancies has provided key insights into their ontogeny, including their cell(s) of origin, the role and nature of antigenic stimulation in tumour development and evolution; moreover, in some diseases, especially CLL,
IGHV gene SHM status has prognostic and predictive value
[37].
While the majority of cases of HCLc have mutated
IGHV genes, 17–20% are unmutated using a 98% cut-off and <5% have completely unmutated
IGHV genes, with 100% identity to the germline. Compared with the normal B-cell repertoire
[38], there is biased usage of the
IGHV3-21, IGHV3-30, IGHV3-33, and
IGHV4-34 genes, each found in 7–10% of cases, with preferential use of
IGHV3-30 and
IGHV4-34, especially among the unmutated cases. Biased usage of
IGHD genes has also been documented
[39][40][41][42].
While kappa is the most frequently used immunoglobulin light chain in the normal B-cell repertoire and in other B-cell tumours, HCLc is associated with preferential use of lambda light chains, resulting in an inverted Igκ:Igλ ratio (0.7:1). The explanation for this is unclear but may derive from secondary IG light chain gene rearrangements as part of receptor editing, a physiological process leading to the pairing of the authentic heavy chain with a novel light chain as a means to alleviate intense autoreactivity.
While there is much less diversity within the light chain, compared with the heavy chain repertoire, there is evidence of biased usage in HCLc, as virtually all cases that express lambda light chains utilise the IGLJ3 gene. All of the above features support a role for antigen selective pressure in tumour ontogeny. Moreover, the presence of intra-clonal diversification within the clonotypic IG genes indicates that ongoing SHM occurs post-transformation likely in a context of continuous interactions with antigen(s).
An unusual feature of HCLc is the expression of multiple
IGH isotypes on the cell surface, documented in 40% to over 80% of cases. Single-cell analysis has confirmed that this phenomenon is attributable to the expression of multiple isotypes in individual cells rather than to clonal heterogeneity
[43]. The expression of multiple CH isotypes, including IgM with IgG or IgA, has also been reported in HCLv and SDRPL, although it has not been demonstrated if they are expressed in single cells. If so, this might point to a microenvironmental factor.
A further anomalous immunogenetic feature of HCLc is an increased incidence of cells expressing both IG kappa and lambda light chains. Dual expression of IG K and L light chains is rare in health, documented in only 0.2–0.5% of B cells from five normal controls
[44]. The functional consequences of dual kappa and lambda light-chain expression are still unknown. That notwithstanding, evidence exists that compromised allelic exclusion leading to dual kappa/lambda expression might allow autoreactive cells to avoid clonal deletion, a mechanism described by the term receptor dilution
[45].
2.6. Biological Implications: Cell of Origin
The stem cell origin of the clonal
BRAFV600E mutation in HCLc, together with the induction of a lethal haematopoietic disorder with features of HCLc in
BRAFV600E mice
[26], are consistent with the role of
BRAFV600E as an early/initiating event in hairy cell leukemogenesis. However, there remains uncertainty about the nature of the mature B-cell population(s) expanded in HCLc and whether additional genetic and/or epigenetic alterations or a suitable TME are required to give rise to mature HCL cells. Dysregulation of the G1 phase of the cell cycle is a common finding in HCLc, but as yet, no subsequent genomic final transforming event has been discovered.
Data relevant to determining the COO in HCLc include the following:
This phenotype also delineates a subset of normal B cells present in blood and splenic red pulp but rarely in lymph nodes. CD11c+, Tbet+ B cell numbers increase with age and are expanded in conditions associated with chronic antigenic stimulation such as infections with human immunodeficiency virus and malaria, and in autoimmune diseases such as SLE. However, the CD11c+ Tbet+ phenotype does not, by itself, identify a distinct B-cell population, nor a specific B-cell lineage, and can be found in activated naïve B cells and in memory B cells believed to be generated through follicular or extra-follicular maturation pathways [51][52][53][54][55][56][57]. It would be interesting to analyze the transcriptomic and methylation data in HCLc using normal splenic CD11c+ Tbet+ CD27- cells rather than CD27+ memory B cells as the comparator. However, currently, and as in CLL despite extensive studies [58], the COO of HCLc remains enigmatic.
2.7. Clinical Implications of Genetic Features
2.7.1. Diagnosis
The initial description of
BRAFV600E in HCLc failed to find the same mutation in 195 cases of other mature B-cell tumours including CLL, follicular lymphoma, DLBCL, and other splenic lymphomas
[9]. However, subsequent screening of larger cohorts of CLL and myeloma for both
BRAFV600E and other
BRAF hotspot mutations has identified a low incidence of predominantly subclonal V600E and non
V600E BRAF mutations, usually associated with a poorer outcome
[59][60][61][62][63].
Whilst a confident diagnosis of HCLc can be made without knowledge of the
BRAF mutation status, the specificity of the
BRAFV600E mutation for HCLc among splenic lymphomas is valuable when there is diagnostic uncertainty, and its presence underpins the use of targeted inhibitors. If it emerges that widely available diagnostic criteria are unable to distinguish
BRAFV600E HCLc from
BRAFWT HCLc, this would provide an additional rationale for
BRAFV600E mutation screening. Allele-specific PCR performed on blood or marrow aspirate samples has superseded less sensitive molecular techniques such as Sanger sequencing, pyrosequencing, or melting curve analysis
[64]. Digital, droplet PCR has comparable specificity and superior sensitivity to QT–PCR and is a potential method for MRD analysis
[65]. Immunohistochemistry (IHC) using a
BRAFV600E-specific antibody is an alternative method suitable for bone marrow trephine or other tissue sections, with comparable sensitivity and specificity to allele-specific PCR. Next-generation sequencing (NGS) also has high sensitivity but, currently, also has higher costs and longer turnaround time, compared with allele-specific tests
[66].
2.7.2. Prognostic Significance of IGHV Gene Somatic Hypermutation Status
The clinical significance of
IGHV gene SHM status in HCLc has been evaluated in two studies with discordant results. In a trial of single-agent cladribine in 58 previously untreated patients, all expressing annexin A1, failure to respond was observed in 5/6 patients with unmutated
IGHV genes using a 98% cut-off value, only one of whom used
IGHV4-34. Bulky splenomegaly, leucocytosis, and
TP53 abnormalities were present in four, three, and two of the five cases, respectively
[40].
In a cohort of 62 patients with HCLc and 20 with HCLv diagnosed according to the WHO 2008 criteria
[67],
IGHV4-34 was used in 6 (10%) of HCLc and 8 (40%) of HCLv cases, respectively, and was unmutated in all but 1 case, using a 98% cut-off value. A suboptimal response to first-line treatment with cladribine was seen in 4/6
IGHV4-34 HCLc positive cases, compared with 4/56
IGHV4-34 negative cases. A worse response was also seen in
IGHV4-34 positive HCLv cases, suggesting that outcome was more closely related to
IGHV4-34 status than to whether or not patients had HCL or HCLv. However, many of the HCLc cases were
BRAFV600E negative
[68].
2.7.3. BRAFV600E as a Therapeutic Target
The purine nucleoside analogues (PNAs), pentostatin, and cladribine remain the current treatment of choice for first-line therapy of HCLc. However, PNAs may cause short-term myelosuppression, with an increased risk of infection and an increased risk of secondary malignancies, and approximately 50% of patients eventually relapse. Single-agent vemurafenib or dabrafenib resulted in high overall response rates without minimal/measurable residual disease (MRD) negativity in relapsed/refractory HCL, but the median relapse-free survival in responders was less than 1 year
[69][70]. In contrast, a phase II study of vemurafenib plus rituximab achieved a CR rate of 87%, of whom 65% were MRD negative and with relapse-free survival of 85% at a median follow-up of 34 months
[71].
2.7.4. Genomic Abnormalities as Predictors of Drug Resistance
Targeted mutational and copy number analysis showed no difference in the pattern of genomic abnormalities between treatment naïve cases and those refractory to a PNA
[11]. Serial samples from two HCL-c cases tested both at diagnosis and relapse post-PNA therapy, revealed two additional subclonal mutations of
BCOR (BCORE1430X) and
XPO1 (XPO1E571K) in one case, while the second case remained genomically stable
[72]. However, there is no clear evidence to suggest that genomic mutations confer resistance to PNAs in HCLc.
Of 13 evaluable HCLc cases treated with vemurafenib, 6 showed persistence of ERK phosphorylation in bone marrow cells, suggesting that, in at least some patients, the growth of HCL cells remains dependent on MEK–ERK signalling, likely reactivated through mechanisms bypassing
BRAF inhibition by vemurafenib. In support, targeted sequencing of 300 genes performed in one patient who was refractory to vemurafenib showed two separate activating subclonal
KRAS mutations at relapse
[71].
A further case with vemurafenib resistance had heterozygous deletions of
BRAF, NF1, NF2, and
TP53 and subclonal mutations in
CREBBP and
IRS1 in a pretreatment sample.
NF1 and
NF2 encode tumour suppressors that have been experimentally implicated in RAF inhibitor resistance in epithelial cancer cells
[73] and downregulation of either or both Nf1 or Nf2 in Ba/F3 cells stably expressing
BRAFV600E conferred vemurafenib resistance in vitro
[11].
Seven distinct activating mutations in
KRAS and two mutations in
MAP2K1 were detected in the relapse sample of a patient resistant to a PNA and vemurafenib plus rituximab. Allele frequencies were consistent with the parallel, convergent evolution of multiple clones with
KRAS mutations appearing before
MAP2K1 mutations. Treatment with MEK inhibitor cobimetinib in combination with vemurafenib resulted in significant clinical and haematological improvement, associated with suppression of mutant allele frequencies for
BRAF, KRAS, and
MAP2K1 mutations and of ERK activity
[74].
Elucidating the mechanisms of resistance to
BRAF inhibitors in solid tumours, especially melanoma, is an area of intensive investigation. In addition to the selection of genomic mutations such as mutations of
RAS or
MAP2K1/MEK1 or of drug-tolerant persister cells, it is increasingly recognised that tumour cells may undergo non-genetic adaptive changes such as metabolic reprograming or reversion to a progenitor cell phenotype which result in drug resistance. It remains to be seen whether such adaptive changes will emerge in HCLc, a tumour with significantly less genomic complexity and instability
[75][76][77][78][79].
3. Conclusions and Future Studies
The major focus was the key role that the discovery of the almost ubiquitous clonal BRAFV600E mutation has played in understanding the biology of HCLc and its importance both in differential diagnosis and as a therapeutic target. However, there remain many unanswered questions regarding the diagnosis and biology of both HCLc and, particularly, HCLv and SDRPL. Of greater clinical importance is an unmet need for potentially curative non-chemotherapeutic regimens for HCLc and more effective treatments for HCLv which additional genetic data may help to resolve.
While the finding of a
BRAFV600E mutation in HCLc unequivocally identifies a disorder with largely uniform laboratory and clinical features, methylome and clinical course, the pathogenesis of less frequent features such as skeletal involvement, found in 3% of cases
[80], and a propensity to autoimmune disease
[81] remain unexplained. Additionally, there is still much to learn about the incidence, biology, and optimal management of cases with a typical HCLc phenotype that lacks the
BRAFV600E mutation or another mechanism for
BRAF upregulation.
It is also unlikely to be coincidental that BRAF WT HCLc cases display enrichment for IGHV4-34 gene usage, frequently accompanied by activating MAP2K1 mutations, and that these two features are also found in a subset of cases with HCLv, raising questions about the inter-relationship between these two patient groups. If IGHV4-34-positive, MAP2K1-mutated cases of HCLc and HCLv do exhibit the typical phenotypes of HCLc and HCLv, respectively, what might account for the differences between the two phenotypes?
There is also uncertainty about the relationship between HCLv and SDRPL, given their many overlapping features and the current absence of disease-defining genetic abnormalities. The absence of reports of the rare cases of SDRPL with progressive disease acquiring typical features of HCLv such as TP53 abnormalities or prominent nucleoli would suggest they are not simply different stages of a single disease.
These uncertainties are, in large part, a consequence of the rarity of these disorders, the lack of cell lines and animal models, and the difficulty in obtaining tumour cells, especially in HCLc, where the circulating tumour cell count is usually low, bone marrow aspiration is unsuccessful, and splenectomy rarely performed
[82][83]. This is reflected in the lack of genomic data on HCLc and especially HCLv and SDRPL, compared with that available in the more common B-cell tumours, such that the published genomic landscapes are unlikely to reflect the full range or true incidence of CNAs and mutations present in all three disorders.
New biological insights are likely to require studies in larger multi-institution patient cohorts, together with the application of newer technologies such as WGS, and transcriptomic and epigenetic analyses, both at the bulk and single-cell levels, comparing data from tumour cells with that from normal splenic B-cell subsets.
It is conceivable that these studies, in conjunction with a more detailed analysis of the TME, may lead to the identification of new disease subsets within or spanning the current diagnoses of BRAF WT HCLc, HCLv, and SDRPL, offer new insights into their cells of origin, and give rise to a more genetically based classification, offering more precise diagnostic and prognostic features and targeted therapies.



