1. Introduction
Loop-mediated isothermal amplification (LAMP) is conducted in the presence of
Bacillus stearothermophylus (
Bst) DNA polymerase, deoxyribonucleotide triphosphate (dNTP), and 4–6 specific primers (inner and outer primers) that recognize 6–8 specific regions
[1]. The
Bst DNA polymerase removes the need for high thermocycling because of its activity at 50–70 °C, and the DNA polymerase is less sensitive to food-origin inhibitors
[2][3]. The use of two outer primers (forward outer primer; F3, and backward outer primer; B3), two inner primers (forward inner primer; FIP, and backward inner primer; BIP), with additional loop primers (forward loop primer; LF, and backward loop primer; LB) also allows sequence-specific detection, improves specificity, and accelerates assay specificity to amplification targets
[4]. However, although many long primers increase the reaction yield, a risk of primer dimerization owing to nonspecific interactions simultaneously exists
[5][6]. To prevent primer dimerization, adding dimethyl sulfoxide (DMSO) and betaine is used as a strategy to decrease nonspecific interactions, thereby giving stability to oligonucleotides
[7]. The use of additional compounds can amplify nucleic acids without non-specific amplification, and exponential nucleic acid amplification is achieved while forming loops within 40–60 min at a constant temperature of 60–65 °C
[8]. Then, the amplified product can be detected using turbidity measurement, gel electrophoresis, colorimetry, electrochemiluminescence, lateral flow assay (LFA), and real-time monitoring
[9][10].
Recombinase polymerase amplification (RPA) is conducted while a recombinase protein UvsX from T4-like bacteriophages forms a complex with primers in the presence of ATP and a crowding agent, such as polyethylene glycol (PEG)
[11]. The crowding agent prevents spontaneous recombinase-primer degradation, thereby allowing amplification to begin. It also enhances the amplification efficiency by improving catalytic activity of the enzyme
[12][13]. Therefore, using long primers (up to 45 nucleotides) can form secondary structures and potential primer artifacts; recommended length of RPA primers is 30–35 bases
[11][14]. RPA is conducted at relatively low and constant temperatures of 37–42 °C for 20–40 min, and detection of amplicons are performed through gel electrophoresis, flocculation assay detection, LFA, electrochemical assay, chemiluminescent assay, and silicon microring resonator (SMR)-based photonic assay, and real-time monitoring
[15][13][16].
HDA reacts with helicase, two primers, and two accessory proteins; methyl-directed mismatch repair (MutL), and single-stranded DNA (ssDNA) binding (SSB) proteins that stimulate UvrD helicase activity above ten-fold
[17][18]. During the HDA replication process, the helicase unwinds double-stranded DNA (dsDNA) for the denaturation. In this step, the accessory proteins are required to bind and stabilize the ssDNA for the prevention of recombination of the complementary strand, thereby allowing primer hybridization
[19]. The HDA reaction is divided into two systems: the mesophilic form of HDA (mHDA) and the thermophilic form of HDA (tHDA). The mHDA reacts at a medium temperature of 37 °C using UvrD helicase/Exo-Klenow polymerase; the tHDA reacts at a higher temperature (60–65 °C) using a thermostable Tte-UvrD helicase/
Bst DNA polymerase
[20]. Compared to mHDA, tHDA has a higher sensitivity and efficiency, and simplifies the reaction because it does not need MutL and SSB proteins to stabilize the DNA sequence
[21]. The magnesium ion, which serves as a cofactor for the helicase and polymerase, is also used in HDA to increase the enzyme activity, thereby making the enzymes compatible with structurally modified primers
[5][22]. Subsequently, amplification products are detected using gel electrophoresis, colorimetry assay, LFA, and real-time monitoring
[21].
Nucleic acid sequence-based amplification (NASBA) targets 16s rRNA genes or mRNA transcripts for bacterial detection, enabling the analysis of bacterial viability
[23][24]. NASBA amplifies single-stranded RNA using two primers and three enzymes (avian myeloblastosis virus reverse transcriptase (AMV-RT), RNase H, and T7 DNA-dependent RNA polymerase (DdRp))
[25]. While AVM-RT generates complementary complementary DNA by extending primers, dsDNA is formed through RNase H. However, T7 DdRP recognizes the exposed T7 promoter of the dsDNA and initiates transcription to initiate the reaction
[25]. Since NASBA enzymes are heat labile, amplifications can be performed at a relatively low temperature, with optimal conditions of 41 °C for 1.5–2 h
[26]. The low reaction temperature of NASBA, however, can produce to result in false-positive results because of nonspecific primer interactions. Nevertheless, adding DMSO and betaine can prevent this limitation
[27][28]. Subsequently, amplification products are detected using gel electrophoresis, enzyme-linked immunosorbent assay (ELISA), enzyme-linked gel assay, electrochemiluminescent (ECL), and real-time monitoring with molecular beacons
[5][29]. When the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system and NASBA are combined, RNA can be detected with high sensitivity through the NASBACC (NASBA-CRISPR Cleavage) system
[30].
RCA has high specificity and sensitivity targeting RNA and DNA
[31]. The RCA reaction is conducted using DNA/RNA polymerase (phi29 DNA polymerase or T7 RNA polymerase), short DNA or RNA linear single-stranded primer, circle template, and ligase
[32]. DNA/RNA polymerase produces a long single-stranded RCA product (RCAP) complementary to a circular template. Unique oligonucleotide padlock probes (PLP) and T4 DNA ligase or special ssDNA ligase synthesize bacterial single- or double-stranded RNA/DNA templates into single-stranded circular DNA
[5]. The various modified RCA systems have been developed for efficient amplification: a linear-RCA (LRCA) or an exponential-RCA (ERCA), such as multiply-primed RCA (MPRCA), hyperbranched RCA (HRCA), and primer-generation RCA (PG-RCA)
[25]. The saltatory RCA (SRCA), a simplified form of RCA, has been developed, requiring ligase and PLP for cyclization
[33][34]. RCA generally reacts for 1–1.5 h at 30–65 °C depending on the reaction system, and amplification products are detected using gel electrophoresis, colorimetry, and real-time monitoring
[25].
MDA randomly and massively amplifies single-cell genomic DNA and is compatible with whole genome amplification (WGA)
[35][36]. MDA is conducted using modified random hexamer primers, phi29 DNA polymerases (strand-displacing DNA polymerase from bacteriophage Ø29), denatured template DNA, and dNTPs
[37]. The modified random hexamer primers eliminate the need to design target-specific primers are designed to anneal to random areas on each strand of the target DNA, thereby forming hyperbranched intermediates and dsDNA amplicons after exponential amplification
[38]. The phi29 DNA polymerase enables amplification at a relatively low temperature (typically 30 °C) because of its high strand displacement activity; it has a higher replication fidelity and lower error rate than
Taq DNA polymerases and
Bst DNA polymerases
[39][40]. Furthermore, the addition of PEG to the reaction for high-efficiency MDA causes molecular crowding, which enables sensitive allele detection in multiplex short tandem repeat genotyping
[41].
Schematic diagram and summary of isothermal amplification techniques are shown in Figure 1 and Table 1.
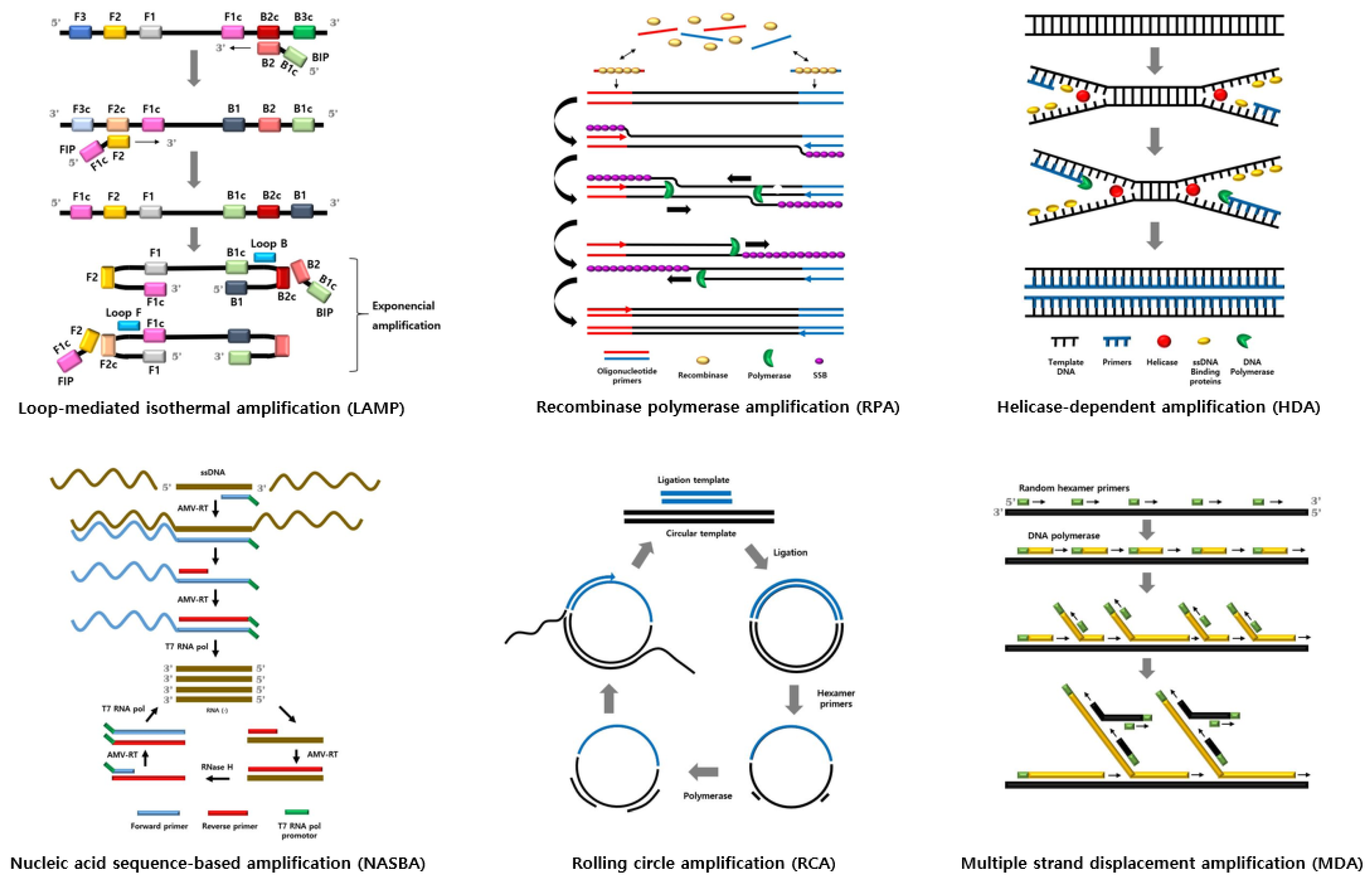 Figure 1.
Figure 1. Schematic diagram of isothermal amplification techniques.
Table 1. Summary of comparison among various isothermal amplification methods.
| Isothermal Amplification Methods |
Number of Primers |
Number of Enzymes |
Pre-Heating |
Working Temperature (°C) |
Reaction Time (min) |
Target Template |
Amplicon |
Resistance to Inhibitor |
Reference |
| LAMP |
4–6 |
1 |
No |
60–65 |
40–60 |
DNA |
DNA |
High |
[8] |
| RPA |
2 |
2 |
No |
37–42 |
20–40 |
DNA |
DNA |
Low |
[15] |
| HDA |
2 |
1 (mHDA), 3 (tHDA) |
No |
37 (mHDA), 60–65 (tHDA) |
100–120 |
DNA |
DNA |
High |
[21] |
| NASBA |
2 |
2–3 |
Yes |
41 |
90–120 |
RNA |
RNA, DNA |
Low |
[26] |
| RCA |
1 |
1 |
Yes |
30–65 |
60–90 |
Circular DNA |
DNA |
Low |
[25] |
| MDA |
Random hexamer primers |
1 |
No |
35 |
270 |
Circular or linear DNAs |
Ramified double–stranded DNAs |
High |
[42] |
Abbreviations used in the table: LAMP, loop-mediated isothermal amplification; RPA, recombinase polymerase amplification; HDA, helicase-dependent amplification; mHDA, mesophilic form of HDA; tHDA, thermophilic form of HDA; NASBA, nucleic acid sequence-based amplification; RCA, rolling circle amplification; MDA, multiple strand displacement amplification.
2. Inhibitors Originating from Isothermal Amplification Processes
Various inhibitors repress the isothermal amplification technique for detecting bacteria, including food-borne pathogens. These inhibitors originate from amplification or detection processes and the food metrix (Table 2).
Table 2. Inhibitors from the isothermal amplification reaction process.
| Reaction Process |
Inhibitors |
Alleviation Strategies for Inhibition |
Amplification Methods |
Reference |
| Sample preparation and DNA extraction |
Residual food metrix |
Use the nucleic acid sample after dilution |
NASBA |
[43] |
| CTAB used as extraction buffer |
Use direct PCR buffers |
RPA |
[44] |
| Nucleic acid amplification |
Concentration |
Magnesium ions |
Increase the concentration of magnesium ions |
tHDA |
[45][46] |
| Add betaine, DMSO, and sorbitol to the reaction mixture |
[17] |
| Use 4–6 mM MgSO4, which is the optimal concentration for magnesium ions |
RCA |
[47] |
| Primer |
Optimize concentration of primer |
RPA |
[11] |
| Multi-RPA |
[48] |
| Template or background DNA |
Treat RNase A with pasteurization and 15 min incubation process before nucleic acid extraction |
Real-time NASBA |
[49] |
| Add the primer stability enhancer to the primer and beacon mixture |
NASBA |
[50] |
| Temperature |
Temperature fluctuations |
Optimize reaction temperature |
MDA |
[51] |
| Heat denaturation |
Substitute alkaline denaturation |
[52] |
| Detection method |
Colorimetric detection |
SYBR Green I |
Add fluorescent dyes after amplification |
LAMP |
[53] |
| Use wax capsules containing the dye, which react after amplification |
[52] |
| PEI |
Add PEI after amplification |
LAMP |
[54] |
| Calcein |
- |
- |
| Electrochemical detection |
Redox active compounds (e.g., MB and Hoechst 33258) |
Use other redox molecules (e.g., osmium redox and RuHex) |
LAMP |
[55] |
| Use voltammeric mode |
[53] |
| Use polydopamine-doped paper disks |
[56] |
Bst DNA polymerase and helicase that enables isothermal amplification require a relatively high concentration of magnesium ions of about 4–8 mM compared to
Taq DNA polymerase
[18]. However, magnesium ions can inhibit molecular amplification depending on the concentration in the reaction mixture
[47]. Murakami et al.
[47] reported that by increasing the background signal amplification in RCA, the concentration of magnesium ions inhibited the signal during target gene amplification. Nb.BsmI, a nicking enzyme, needs magnesium ions to enhance amplification efficiency. Therefore, optimizing magnesium ion concentrations (4–6 mM MgSO
4) and decreasing dNTPs and DNA polymerase concentrations can decrease background signal amplifications. Doseeva et al.
[46] conducted a study to alleviate magnesium ion-dependent inhibition because the concentration of magnesium ions affected the amplification efficiency of tHDA. Results indicated that as the concentration of magnesium ions and that of the dATPs improved, the signal-to-noise ratio increased 1.5–2.0 times. It was also observed that the optimal concentrations of MgSO
4, dATP, and dNTP were 4, 3, and 0.4 mmol/L, respectively. Additionally, betaine, DMSO, and sorbitol, which help the combined effect of magnesium ions and dATP during molecular amplification, were added to increase the efficiency and specificity of amplification in tHDA.
The inappropriate concentration of primers can inhibit molecular amplification processes. Thus, SSB proteins either inhibit the strand exchange activity of recombinase T4 UvsX or compete with recombinase proteins in RPA
[57]. Additionally, primers for one target can inhibit the amplification of another target
[11]. Therefore, it is essential to optimize primer concentration
[11][16].
3. Conclusions
Inhibitors originating from food matrices, DNA extraction reactions and nucleic acid amplification reactions can adversely affect isothermal amplification techniques by inhibiting amplification to detect food-borne pathogens. Nevertheless, due to the complex nature of the food matrix, food-origin inhibitors, including inhibition mechanisms for molecular analysis, have not been fully characterized yet. Additionally, studies on the inhibitory mechanisms of inhibitors obtained during isothermal amplification processes are still insufficient and have not been clearly established. The above-mentioned removal strategies for inhibiting nucleic acid amplification are rather limited and uneconomical because they only target specific inhibitors, and have not been proven to be applicable to various inhibitors. Therefore, further studies on nucleic acid amplification inhibitors and inhibitor removal strategies are needed to detect food-borne pathogens in food using isothermal amplification technologies without inhibition of nucleic acid amplification inhibitors. In the future, these studies will lead to the manufacture of a ready-to-use kit that simultaneously purifies and removes inhibitors. In this case, efficiency is guaranteed for accurate detection of food-borne pathogens in complex food matrices and for the point-of-care testing (POCT) application of these isothermal amplification technologies.
 +1 credit
+1 credit



