Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicholas Alexander Hulett | + 2442 word(s) | 2442 | 2022-02-11 04:20:13 | | | |
| 2 | Camila Xu | -2 word(s) | 2440 | 2022-03-07 04:42:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hulett, N. Glucose Uptake by Skeletal Muscle. Encyclopedia. Available online: https://encyclopedia.pub/entry/20225 (accessed on 23 July 2026).
Hulett N. Glucose Uptake by Skeletal Muscle. Encyclopedia. Available at: https://encyclopedia.pub/entry/20225. Accessed July 23, 2026.
Hulett, Nicholas. "Glucose Uptake by Skeletal Muscle" Encyclopedia, https://encyclopedia.pub/entry/20225 (accessed July 23, 2026).
Hulett, N. (2022, March 04). Glucose Uptake by Skeletal Muscle. In Encyclopedia. https://encyclopedia.pub/entry/20225
Hulett, Nicholas. "Glucose Uptake by Skeletal Muscle." Encyclopedia. Web. 04 March, 2022.
Copy Citation
Skeletal muscle is the primary tissue for maintaining glucose homeostasis through glucose uptake via insulin-dependent and -independent mechanisms. Skeletal muscle is also responsive to exercise-meditated glucose transport, and as such, exercise is a cornerstone for glucose management in people with type 2 diabetes.
type 2 diabetes
insulin resistance
glucose transport
skeletal muscle
1. Introduction
Insulin-mediated skeletal muscle glucose uptake requires a concert of physiological events. Upon ingestion of nutrients, the pancreas senses glucose elevation, leading to glucose-dependent insulin secretion and suppression of glucagon secretion. Insulin must circulate to target tissues to bind and take effect. One aspect of insulin action often underappreciated is insulin-dependent stimulation of endothelial nitric oxide synthase (eNOS). eNOS augments blood flow to insulin-sensitive target organs, including the cardiac and skeletal muscle. As such, insulin actively regulates delivery of itself, nutrients, and oxygen to the skeletal muscle. In the skeletal muscle, glucose and insulin must leave the circulation and traverse the endothelium and extracellular matrix to cross through the cell membrane to stimulate or enter the myocyte. Glucose can enter the skeletal muscle via both insulin-dependent and insulin-independent glucose transporters, including facilitated glucose transporter members (GLUT 1, 3, 4, 5, 8, 10, 11, and 12) [1].
2. Role of Skeletal Muscle in Blood Glucose Regulation
Skeletal muscle plays a principal role in post-prandial glucose regulation. After ingestion, ~80% of glucose is taken up by skeletal muscle via insulin-dependent glucose uptake [2][3]. Insulin-dependent and -independent skeletal muscle glucose disposal requires (1) glucose delivery to the muscle from circulation, (2) glucose traversing the extracellular matrix to the cell membrane, (3) uptake via facilitative glucose transporters either constitutively on the cell membrane or translocated in response to insulin or exercise, and (4) glucose gradient to facilitate glucose transport modulated by intracellular glucose metabolism, as shown in Figure 1.
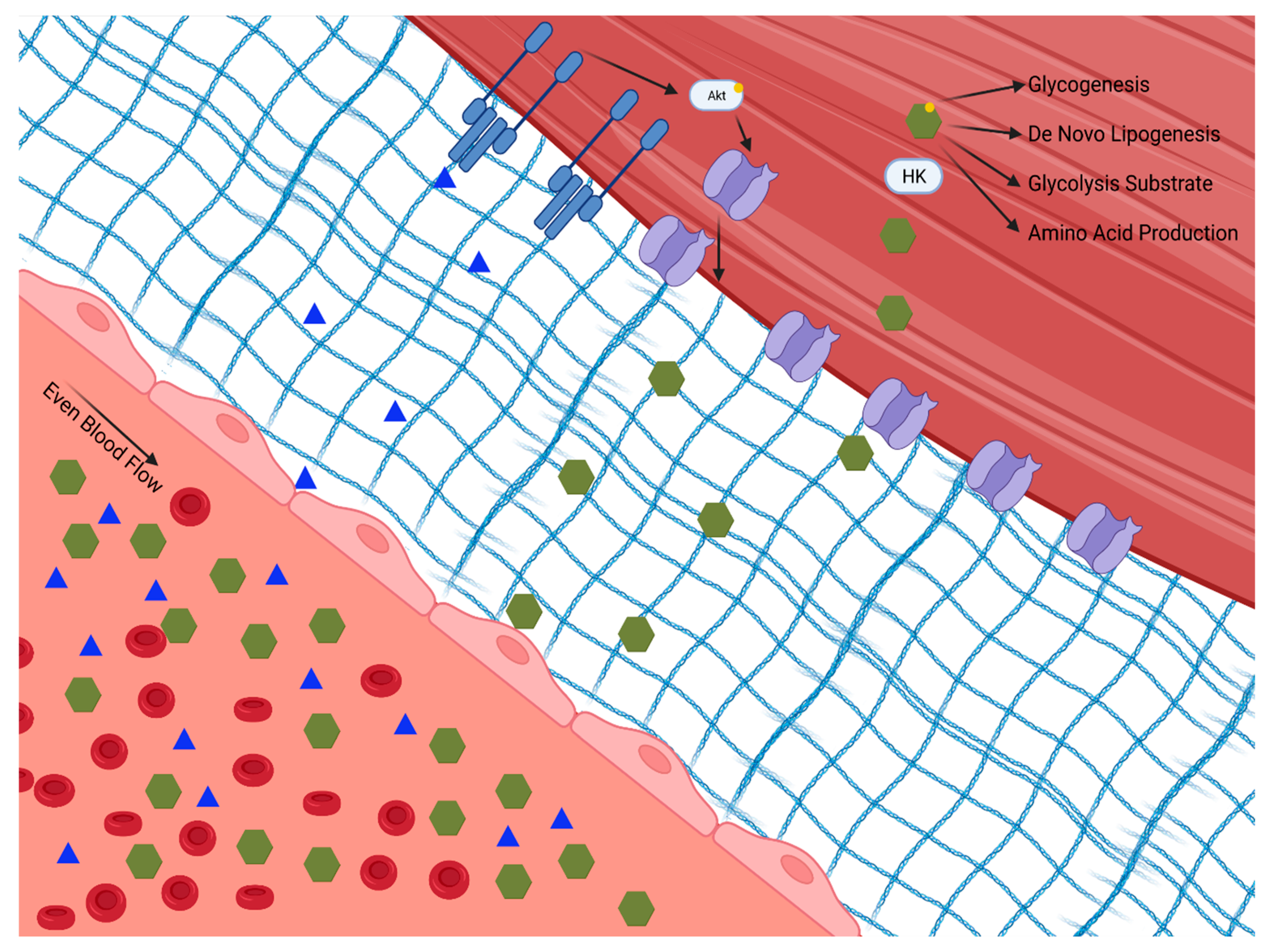 Figure 1. Insulin-dependent and -independent skeletal muscle glucose disposal requires: (1) glucose delivery to the muscle from circulation through the extracellular matrix to the cell membrane; (2) uptake via facilitative glucose transporters either constitutively on the cell membrane or translocated in response to insulin or exercise; and (3) a glucose diffusion gradient to drive glucose into the cell which is modulated by intracellular glucose metabolism. Hexokinase (HK). Phosphokinase B (Akt).
Figure 1. Insulin-dependent and -independent skeletal muscle glucose disposal requires: (1) glucose delivery to the muscle from circulation through the extracellular matrix to the cell membrane; (2) uptake via facilitative glucose transporters either constitutively on the cell membrane or translocated in response to insulin or exercise; and (3) a glucose diffusion gradient to drive glucose into the cell which is modulated by intracellular glucose metabolism. Hexokinase (HK). Phosphokinase B (Akt).The initial step for skeletal muscle glucose clearance is delivery. As such, skeletal muscle blood flow and perfusion play a key role in glucose disposal, which is often overlooked. It is interesting to speculate that tissue delivery is why insulin has evolved to regulate eNOS-dependent vasodilation [4]. The endothelium expresses the insulin and insulin like growth factor 1 (IGF 1) receptors [5]. When these receptors are activated, eNOS is activated via the Phosphokinase B (Akt/PKB) pathway, leading to vasodilation [6]. Experimental disruption of insulin signaling to eNOS, either globally or specifically in the skeletal muscle, lowers whole-body glucose disposal [7]. This mechanism of insulin action appears to be specifically vital in skeletal muscle, as blocking insulin-induced vasodilation in adipose tissue does not impair glucose uptake [8]. Insulin-mediated augmentation of perfusion of the skeletal muscle provides access to insulin, glucose, and oxygen, all of which are essential for glucose clearance and metabolism.
When skeletal muscle is perfused, glucose can be cleared from the circulation through the interstitial space and into the skeletal muscle. The extracellular matrix of healthy skeletal muscle is free of inflammation and fibrosis, allowing for rapid diffusion and transport of insulin and glucose across the endothelium and basement membrane to the skeletal muscle [9]. In the fasted state, the skeletal muscle is exposed to low insulin levels. In this state, glucose transport in skeletal muscle is facilitated via constitutive transporters on the skeletal muscle membrane [10]. After feeding, circulating insulin levels increase, insulin binds to its skeletal muscle receptor and signals GLUT translocation to the membrane. Briefly, insulin binds the insulin receptor causing phosphorylation of the insulin receptor substrate, which then activates the Akt/PKB pathway. Activation of the Akt/PKB pathway triggers the translocation of GLUT4 from the cytosol into the membrane, allowing glucose to move down its concentration gradient into the cell [11]. Of note, there is a complex post-receptor interplay of multiple signaling events in the glucose transport signaling pathway. This process, discovered more than 30 years ago, is now understood in exquisite detail, and has recently been reviewed [12][13][14][15]. GLUT4 is the main GLUT isoform that translocates to the cell membrane with insulin stimulation; however, GLUT 12 has also been shown to embed, as well [16].
Insulin is not unique in its ability to enhance skeletal muscle glucose transport. Contraction of skeletal muscle also signals GLUT4 translocation. Multiple upstream mechanisms including Rac1/actin and Ca2+/calmodulin-dependent protein kinase (CAMK) signaling are activated via muscle contraction [17]. These two signaling networks share downstream key proteins such as AMP-activated protein kinase (AMPK) and PI3K while also relying on novel targets such as TBC1D1/4 [18][19]. While GLUT4 is the most highly expressed glucose transport protein in skeletal muscle, there are other GLUT isoforms with physiological significance, notably GLUT1, which is constitutively present on the surface of skeletal muscle [20]. When GLUT1 is experimentally increased, basal glucose uptake also increases [21]. Glucose has also been shown to stimulate its own uptake through a process known as “glucose effectiveness” [22]. Each GLUT isoform is a facilitated transporter requiring a diffusion gradient to drive glucose into the cell.
Once in the cell, glucose must be utilized both to meet metabolic demands and to maintain the concentration gradient for facilitated transport. Glucose is initially phosphorylated via hexokinase, which traps the glucose intracellularly in the skeletal muscle, where it will succumb to one of four main fates: storage as glycogen, substrate in glycolysis, substrate for protein synthesis via the hexosamine pathway, or substrate in the pentose phosphate pathway [23]. Selection of glucose fate is dependent on the metabolic demands of the cell. In times of low energetic demand, glucose can be stored as glycogen, a highly branched polysaccharide. Glycogen storage is limited in skeletal muscle and therefore regulated by negative feedback on glycogen synthase by glycogen. Phosphorylated glucose can also enter glycolysis, a key ATP-producing pathway which also supplies fuel for the Krebs cycle and oxidative phosphorylation. This pathway is also negatively regulated by its end products, which accumulate during times of low energetic demand. The hexosamine pathway uses an intermediate product of glycolysis, fructose-6-phosphate, to produce nucleotides needed for protein synthesis [24]. Lastly, the pentose phosphate pathway is responsible for nicotinamide adenine dinucleotide phosphate, ribose 5-phosphate, and erythrose-4-phosophate production, which are critical factors for anabolism [25]. Flux through these pathways is driven by the needs of the cell in a dynamic manner. Each of these pathways can become saturated with excess glucose influx.
When skeletal muscle is adequately supplied with nutrient rich blood, has sufficient membrane permeability to glucose, and can maintain a diffusion gradient by storage and metabolism, large amounts of glucose can be cleared, maintaining systemic carbohydrate homeostasis. Skeletal muscle is a primary target of systemic glucose disposal; thus, interference in skeletal muscle glucose transport is sufficient to induce whole-body insulin resistance [26][27]. Loss of skeletal muscle glucose uptake is associated with abnormal carbohydrate metabolism, T2D, and associated with the development of atherogenic dyslipidemia [28]. While debated, there is evidence in humans that skeletal muscle acquires insulin resistance before adipocytes and hepatocytes [28].
3. Changes in Skeletal Muscle Physiology in Type 2 Diabetes
Skeletal muscle in people with T2D demonstrates diverse pathological changes impacting the delivery, uptake, and metabolism of glucose. The ability of skeletal muscle to respond to insulin is a primary factor in the pathophysiology associated with T2D. This phenotype likely has several converging mechanisms of action whose relative contributions remain contentious. Briefly, in individuals with a genetic predisposition for T2D, chronic nutrient overload, physical inactivity, and subsequent obesity lead to excess and ectopically stored lipid. The species of lipid, the cellular location, and turnover rates all influence insulin sensitivity [29]. For example, the increased diacylglycerol concentration within skeletal muscle associated with overnutrition and sedentary behavior leads to inflammation, oxidative damage, and fibrosis, reducing skeletal muscle’s ability to respond to demand [30]. Alternative activation of serine/threonine kinases and subsequent decreases in insulin receptor and IRS-1 tyrosine phosphorylation have also been implicated in disruptions of insulin signaling [31]. The skeletal muscle adapts (or maladapts) to chronic nutrient excess with changes in vascular structure and function—including endothelial dysfunction and accumulation of matrix proteins—and alterations in insulin signaling, and then reshapes the metabolic character, all of which contribute to reduced skeletal muscle glucose uptake, as shown in Figure 2 [32].
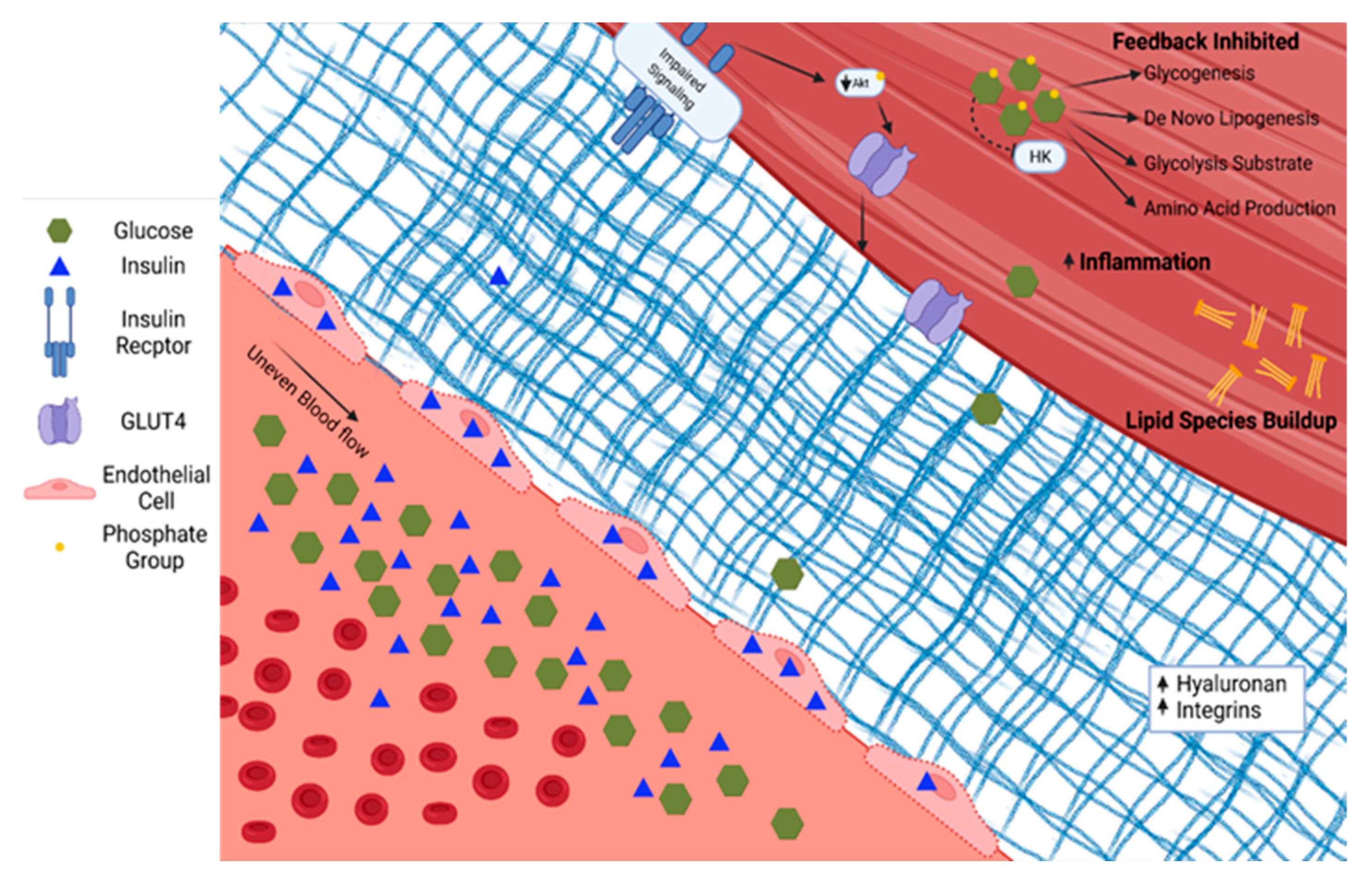 Figure 2. Type 2 diabetes is characterized by increased glucose and insulin in circulation. Insulin accumulates in endothelial cells. The extracellular matrix becomes fibrotic with increased hyaluronan and integrins. Serine/threonine phosphorylation on the insulin receptor and insulin response substrates leads to blunted insulin signaling through PI3K/Akt. The glucose diffusion gradient is limited by elevated intracellular glucose concentrations and allosteric down-regulation of intracellular glucose metabolism. Hexokinase (HK). Phosphokinase B (Akt).
Figure 2. Type 2 diabetes is characterized by increased glucose and insulin in circulation. Insulin accumulates in endothelial cells. The extracellular matrix becomes fibrotic with increased hyaluronan and integrins. Serine/threonine phosphorylation on the insulin receptor and insulin response substrates leads to blunted insulin signaling through PI3K/Akt. The glucose diffusion gradient is limited by elevated intracellular glucose concentrations and allosteric down-regulation of intracellular glucose metabolism. Hexokinase (HK). Phosphokinase B (Akt).An objective here is to highlight a body of data on the contribution of abnormal microvascular structure and function to insulin resistance and hyperglycemia [33]. Microvascular complications are a hallmark of T2D-specific complications, including cardiomyopathy, nephropathy, retinopathy, and neuropathy. Changes in microvascular structure and function are present in the skeletal muscle, heart, brain, and islet in diabetes [34]. These microvascular changes suggest a role of the vasculature of insulin resistant skeletal muscle. The endothelium lining of the vasculature is responsible for sensing and responding to oxygen and nutrients within the blood [35]. As mentioned above, insulin and glucose are both signals to the endothelium to vasodilate to increase skeletal muscle perfusion and associated delivery and disposal of glucose [36]. However, these signals are disrupted with insulin resistance. T2D causes systemic endothelial cell damage through reactive oxygen and nitrogen species with reduced antioxidant potentials, reducing the cell’s ability to respond to stimuli [37]. For example, nitric oxide production is lower in response to insulin in insulin resistance, resulting in improper perfusion of skeletal muscle and subsequent impairments of insulin, glucose, and oxygen delivery [36][38][39]. Insulin resistance can be partially prevented when vasodilators are given, demonstrating the importance of blood flow for insulin action [40].
The endothelium plays a role in skeletal muscle insulin delivery in the context of T2D. There is debate in the field regarding the primary mechanism by which insulin moves from circulation to skeletal muscle. The two main positions are that insulin travels through the endothelial cell via transport proteins or between cells via fluid-phase transport. In support of insulin traveling through the cell is the observation that insulin accumulates in the endothelial cells at five times the concentration compared with circulation, requiring PI-3 kinase and mitogen-activated protein kinase kinase (MEK-kinase) signaling for proper transport to skeletal muscle [36][41]. Additional reports demonstrate that most insulin leaves circulation via fluid-phase transport and is independent of binding in endothelial cells [38][42]. Hyperglycemia associated with insulin resistance negatively affects insulin delivery to skeletal muscle and directly injures the endothelium [43]. Hyperglycemia causes the formation of advanced glycation end products that lead to proinflammatory signaling in the vessel walls and further reduce endothelial function [44].
Glucose and insulin access to the skeletal muscle are also limited by T2D-mediated changes to the perivascular and skeletal muscle extracellular matrix. The extracellular matrix plays a key role in capillary angiogenesis and regression [45] and insulin signaling [46]. In a pro-inflammatory state, as in T2D, skeletal muscle collagen production and turnover is dysregulated. Extracellular matrix remodeling is necessary for new capillary growth. Dysregulation of matrix remodeling can decrease capillary density and also impede the effective transfer of material from the bloodstream to the skeletal muscle. In T2D, there is an expansion of the extracellular matrix and decreased capillary density [9]. Hyaluronan, a chief element of a structural component of the extracellular matrix, is increased in those with T2D. Insulin action improves when hyaluronan expression is experimentally decreased in high fat fed mice [47]. Collagen deposition is also commonly associated with T2D [46]. Greater collagen is associated with lower muscle matrix metallopeptidase 9 activity in skeletal muscle [48]. Integrins, a class of matrix proteins, actively induce insulin resistance. For example, deletion of integrin-linked kinase from the skeletal muscle of mice had no effect on insulin action in lean mice. In contrast, when the same gene was deleted in obese mice, capillaries and insulin action were greater [49]. Manipulation of downstream molecules such as focal adhesion kinase demonstrate similar results [50]. These data demonstrate important barriers to skeletal muscle insulin delivery and glucose transport upstream of direct insulin action in T2D skeletal muscle.
Changes in skeletal muscle insulin signaling and cellular context occur in T2D. For example, GLUT proteins must be present in the membrane for facilitated glucose transport. In the context of T2D, GLUT4 content and translocation to the membrane is impaired due to insulin resistance. Increasingly, the problem of overnutrition and nutrient load on skeletal muscle has come to the forefront of understanding insulin resistance. Chronic overnutrition results in increased glycerol and free fatty acids in circulation. Normally, these nutrients are widely cleared by skeletal muscle and adipose tissue. Free fatty acids can diffuse into the cell or are taken up via a transporter such as CD36 and stored as intramyocellular lipids. However, excess in intramuscular triglyceride stores have been linked by numerous studies to insulin resistance [51][52][53]. When there is buildup of lipids, toxic metabolites such as diacetyl glyceride, ceramide, and acylcarnitine can accumulate. These metabolites act as signaling molecules that alter proteins and signaling cascades involved in insulin signaling, oxidative phosphorylation, and peroxisomal metabolism in skeletal muscle [54]. In addition, many of these lipid species increase the action of protein kinase Cθ, which phosphorylates serine residues of the insulin receptor and IRS, downregulating their action [55]. Similarly, toxic lipids drive inflammatory processes associated with insulin resistance [56][57]. In the context of excess nutrients, a high protonmotive force and NADH/NAD+ ratio occurs in the mitochondria as fuels are oxidized [58]. These conditions, without a constructive means to dissipate the protonmotive force (such as ATP production driven by demand), create a high H2O2 emitting potential [59][60]. The subsequent free radicals are proposed to further exacerbate insulin resistance [32]. The contribution of oxidative stress to insulin resistance is supported by data showing that treating high fat fed mice with a mitochondrial-specific antioxidant preserves insulin sensitivity [61].
Pathologic intracellular changes in skeletal muscle cell metabolism in T2D create a viscous cycle exacerbating insulin resistance in the context of overnutrition. As mentioned above, glucose enters the cell via facilitated transport down a diffusion gradient. To phosphorylate glucose, hexokinase must be present and active, and its product, glucose-6-phosphate, must be further metabolized to prevent allosteric down-regulation. When these processes are saturated, glucose-6-phosphate concentration increases and inhibits hexokinase activity, decreasing glucose transport. The concentration and compartmentalization of hexokinase are changed in T2D. Insulin signaling increases hexokinase [62]. Due to the relative decrease in insulin signaling with T2D, hexokinase expression levels are approximately 80% lower compared with controls [63].
References
- Evans, P.L.; McMillin, S.L.; Weyrauch, L.A.; Witczak, C.A. Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training. Nutrients 2019, 11, 2432.
- Ferrannini, E.; Simonson, D.C.; Katz, L.D.; Reichard, G., Jr.; Bevilacqua, S.; Barrett, E.J.; Olsson, M.; DeFronzo, R.A. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism 1988, 37, 79–85.
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809.
- Laakso, M.; Edelman, S.V.; Brechtel, G.; Baron, A.D. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J. Clin. Investig. 1990, 85, 1844–1852.
- Li, G.; Barrett, E.J.; Wang, H.; Chai, W.; Liu, Z. Insulin at physiological concentrations selectively activates insulin but not insulin-like growth factor I (IGF-I) or insulin/IGF-I hybrid receptors in endothelial cells. Endocrinology 2005, 146, 4690–4696.
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 2000, 101, 1539–1545.
- Laakso, M.; Edelman, S.V.; Brechtel, G.; Baron, A.D. Impaired insulin-mediated skeletal muscle blood flow in patients with NIDDM. Diabetes 1992, 41, 1076–1083.
- Emanuel, A.L.; Meijer, R.I.; Muskiet, M.H.; van Raalte, D.H.; Eringa, E.C.; Serne, E.H. Role of Insulin-Stimulated Adipose Tissue Perfusion in the Development of Whole-Body Insulin Resistance. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 411–418.
- Ruggiero, A.D.; Davis, A.; Sherrill, C.; Westwood, B.; Hawkins, G.A.; Palmer, N.D.; Chou, J.W.; Reeves, T.; Cox, L.A.; Kavanagh, K. Skeletal muscle extracellular matrix remodeling with worsening glycemic control in nonhuman primates. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R226–R235.
- Wasserman, D.H. Four grams of glucose. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E11–E21.
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871.
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381.
- Cushman, S.W.; Wardzala, L.J. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell: Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 1980, 255, 4758–4762.
- Suzuki, K.; Kono, T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. USA 1980, 77, 2542–2545.
- Wardzala, L.J.; Cushman, S.W.; Salans, L.B. Mechanism of insulin action on glucose transport in the isolated rat adipose cell. Enhancement of the number of functional transport systems. J. Biol. Chem. 1978, 253, 8002–8005.
- Stuart, C.A.; Howell, M.E.; Zhang, Y.; Yin, D. Insulin-stimulated translocation of glucose transporter (GLUT) 12 parallels that of GLUT4 in normal muscle. J. Clin. Endocrinol. Metab. 2009, 94, 3535–3542.
- Wright, D.C.; Hucker, K.A.; Holloszy, J.O.; Han, D.H. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 2004, 53, 330–335.
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017.
- de Wendt, C.; Espelage, L.; Eickelschulte, S.; Springer, C.; Toska, L.; Scheel, A.; Bedou, A.D.; Benninghoff, T.; Cames, S.; Stermann, T.; et al. Contraction-Mediated Glucose Transport in Skeletal Muscle Is Regulated by a Framework of AMPK, TBC1D1/4, and Rac1. Diabetes 2021, 70, 2796–2809.
- Handberg, A.; Kayser, L.; Hoyer, P.E.; Vinten, J. A substantial part of GLUT-1 in crude membranes from muscle originates from perineurial sheaths. Am. J. Physiol. 1992, 262 Pt 1, E721–E727.
- Hansen, P.A.; Wang, W.; Marshall, B.A.; Holloszy, J.O.; Mueckler, M. Dissociation of GLUT4 translocation and insulin-stimulated glucose transport in transgenic mice overexpressing GLUT1 in skeletal muscle. J. Biol. Chem. 1998, 273, 18173–18179.
- Ader, M.; Ni, T.C.; Bergman, R.N. Glucose effectiveness assessed under dynamic and steady state conditions. Comparability of uptake versus production components. J. Clin. Investig. 1997, 99, 1187–1199.
- Bouche, C.; Serdy, S.; Kahn, C.R.; Goldfine, A.B. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 2004, 25, 807–830.
- Love, D.C.; Hanover, J.A. The hexosamine signaling pathway: Deciphering the “O-GlcNAc code”. Sci. STKE 2005, 2005, re13.
- Meijer, A.E. The pentose phosphate pathway in skeletal muscle under patho-physiological conditions. A combined histochemical and biochemical study. Prog. Histochem. Cytochem. 1991, 22, 1–114.
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928.
- DeFronzo, R.A. and D. Tripathy, Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163.
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594.
- Bergman, B.C.; Goodpaster, B.H. Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity. Diabetes 2020, 69, 848–858.
- Lark, D.S.; Wasserman, D.H. Meta-fibrosis links positive energy balance and mitochondrial metabolism to insulin resistance. F1000Resarch 2017, 6, 1758.
- Petersen, K.F.; Shulman, G.I. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 11G–18G.
- Lark, D.S.; Fisher-Wellman, K.H.; Neufer, P.D. High-fat load: Mechanism(s) of insulin resistance in skeletal muscle. Int. J. Obes. Suppl. 2012, 2 (Suppl. 2), S31–S36.
- Abushamat, L.A.; McClatchey, P.M.; Scalzo, R.L.; Schauer, I.; Huebschmann, A.G.; Nadeau, K.J.; Liu, Z.; Regensteiner, J.G.; Reusch, J.E.B. Mechanistic Causes of Reduced Cardiorespiratory Fitness in Type 2 Diabetes. J. Endocr. Soc. 2020, 4, bvaa063.
- Faselis, C.; Katsimardou, A.; Imprialos, K.; Deligkaris, P.; Kallistratos, M.; Dimitriadis, K. Microvascular Complications of Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2020, 18, 117–124.
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial Cell Metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244.
- Barrett, E.J.; Eggleston, E.M.; Inyard, A.C.; Wang, H.; Li, G.; Chai, W.; Liu, Z. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia 2009, 52, 752–764.
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820.
- Liu, J.; Liu, Z. Muscle Insulin Resistance and the Inflamed Microvasculature: Fire from Within. Int. J. Mol. Sci. 2019, 20, 562.
- Clerk, L.H.; Vincent, M.A.; Jahn, L.A.; Liu, Z.; Lindner, J.R.; Barrett, E.J. Obesity blunts insulin-mediated microvascular recruitment in human forearm muscle. Diabetes 2006, 55, 1436–1442.
- Wasserman, D.H.; Wang, T.J.; Brown, N.J. The Vasculature in Prediabetes. Circ. Res. 2018, 122, 1135–1150.
- Genders, A.J.; Frison, V.; Abramson, S.R.; Barrett, E.J. Endothelial cells actively concentrate insulin during its transendothelial transport. Microcirculation 2013, 20, 434–439.
- Williams, I.M.; Valenzuela, F.A.; Kahl, S.D.; Ramkrishna, D.; Mezo, A.R.; Young, J.D.; Wells, K.S.; Wasserman, D.H. Insulin exits skeletal muscle capillaries by fluid-phase transport. J. Clin. Investig. 2018, 128, 699–714.
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057.
- Dobi, A.; Rosanaly, S.; Devin, A.; Baret, P.; Meilhac, O.; Harry, G.J.; d’Hellencourt, C.L.; Rondeau, P. Advanced glycation end-products disrupt brain microvascular endothelial cell barrier: The role of mitochondria and oxidative stress. Microvasc. Res. 2021, 133, 104098.
- Marchand, M.; Monnot, C.; Muller, L.; Germain, S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin. Cell Dev. Biol. 2019, 89, 147–156.
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366.
- Han, C.Y.; Subramanian, S.; Chan, C.K.; Omer, M.; Chiba, T.; Wight, T.N.; Chait, A. Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 2007, 56, 2260–2273.
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 2011, 60, 416–426.
- Kang, L.; Mokshagundam, S.; Reuter, B.; Lark, D.S.; Sneddon, C.C.; Hennayake, C.; Williams, A.S.; Bracy, D.P.; James, F.D.; Pozzi, A.; et al. Integrin-Linked Kinase in Muscle Is Necessary for the Development of Insulin Resistance in Diet-Induced Obese Mice. Diabetes 2016, 65, 1590–1600.
- Bisht, B.; Goel, H.L.; Dey, C.S. Focal adhesion kinase regulates insulin resistance in skeletal muscle. Diabetologia 2007, 50, 1058–1069.
- Pan, D.A.; Lillioja, S.; Kriketos, A.D.; Milner, M.R.; Baur, L.A.; Bogardus, C.; Jenkins, A.B.; Storlien, L.H. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 1997, 46, 983–988.
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116.
- Phillips, D.I.; Caddy, S.; Ilic, V.; Fielding, B.A.; Frayn, K.N.; Borthwick, A.C.; Taylor, R. Intramuscular triglyceride and muscle insulin sensitivity: Evidence for a relationship in nondiabetic subjects. Metabolism 1996, 45, 947–950.
- Sachs, S.; Zarini, S.; Kahn, D.E.; Harrison, K.A.; Perreault, L.; Phang, T.; Newsom, S.A.; Strauss, A.; Kerege, A.; Schoen, J.A.; et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E866–E879.
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790.
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105.
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180.
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272.
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581.
- Vogt, C.; Ardehali, H.; Iozzo, P.; Yki-Jarvinen, H.; Koval, J.; Maezono, K.; Pendergrass, M.; Printz, R.; Granner, D.; DeFronzo, R.; et al. Regulation of hexokinase II expression in human skeletal muscle in vivo. Metabolism 2000, 49, 814–818.
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 1999, 341, 240–246.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.8K
Revisions:
2 times
(View History)
Update Date:
07 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No