 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Larissa Anastacio da Costa Carvalho | + 5109 word(s) | 5109 | 2022-02-23 10:58:14 | | | |
| 2 | Vivi Li | Meta information modification | 5109 | 2022-03-07 02:48:50 | | |
Video Upload Options
Melanoma is the most aggressive type of skin cancer. Despite the available therapies, the minimum residual disease is still refractory. Reactive oxygen and nitrogen species (ROS and RNS) play a dual role in melanoma, where redox imbalance is involved from initiation to metastasis and resistance. Redox proteins modulate the disease by controlling ROS/RNS levels in immune response, proliferation, invasion, and relapse. Chemotherapeutics such as BRAF and MEK inhibitors promote oxidative stress, but high ROS/RNS amounts with a robust antioxidant system allow cells to be adaptive and cooperate to non-toxic levels. These proteins could act as biomarkers and possible targets. By understanding the complex mechanisms involved in adaptation and searching for new targets to make cells more susceptible to treatment, the disease might be overcome.
1. Introduction
2. RNS and ROS-Generating Enzymes
2.1. Nitric Oxide Synthase (NOS)

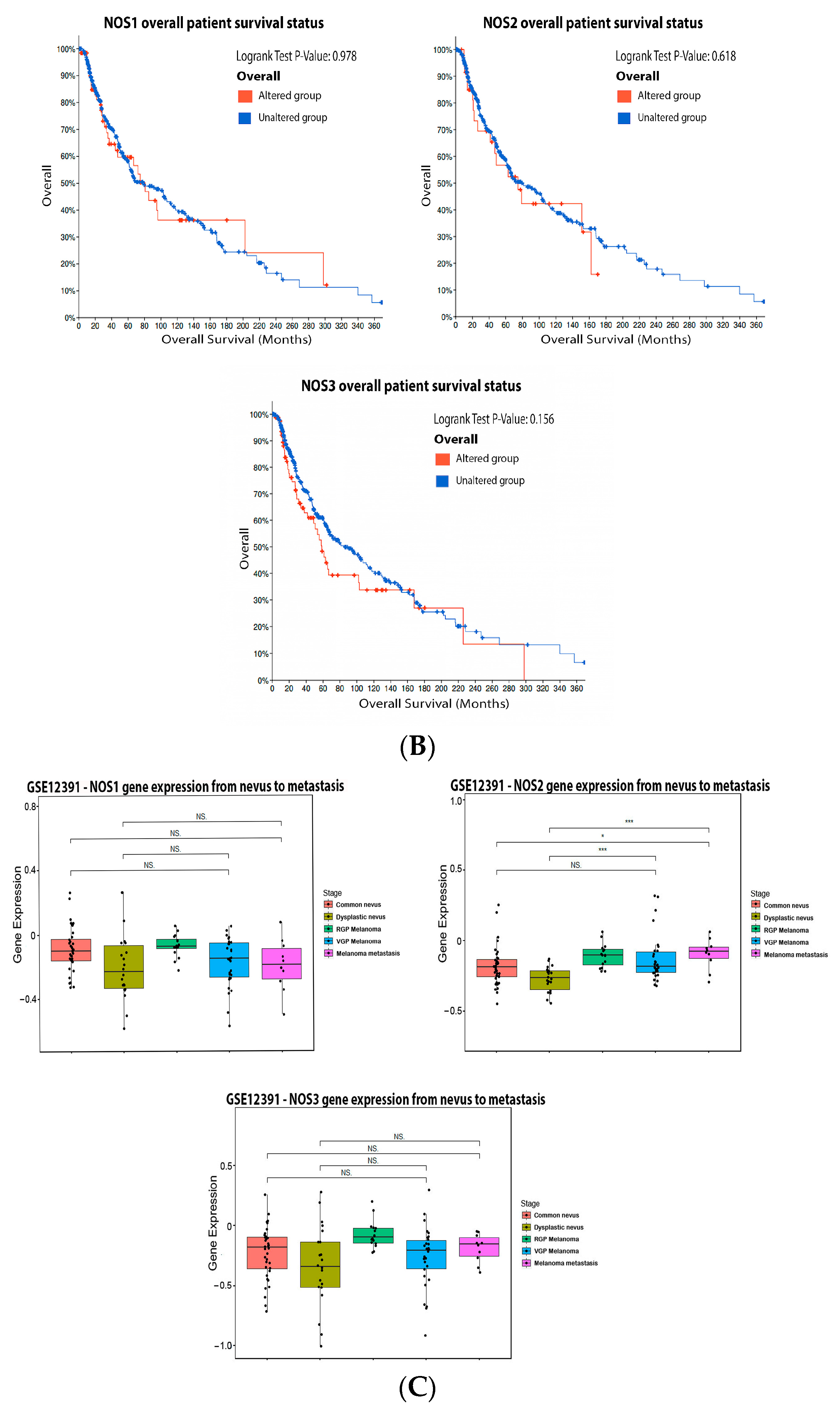
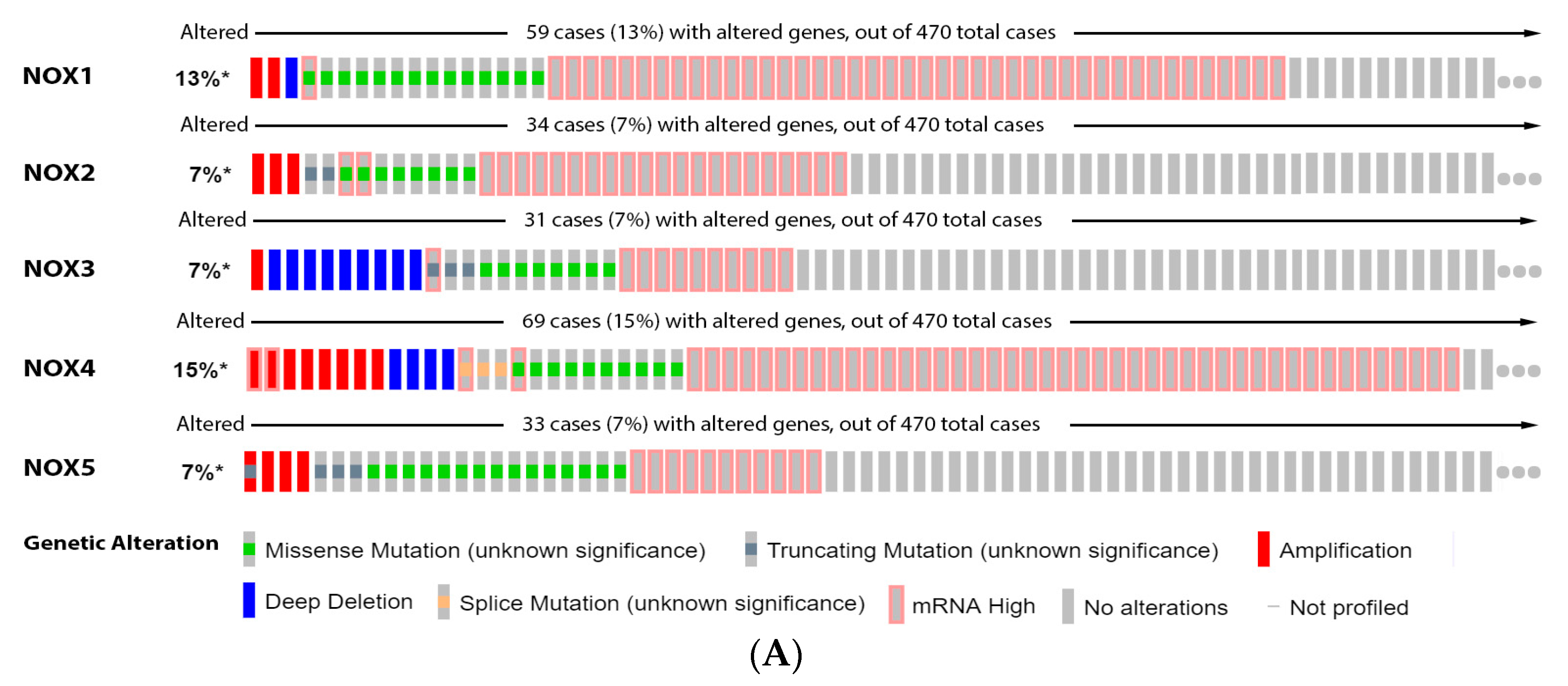
2.2. NADPH Oxidase (NOX)
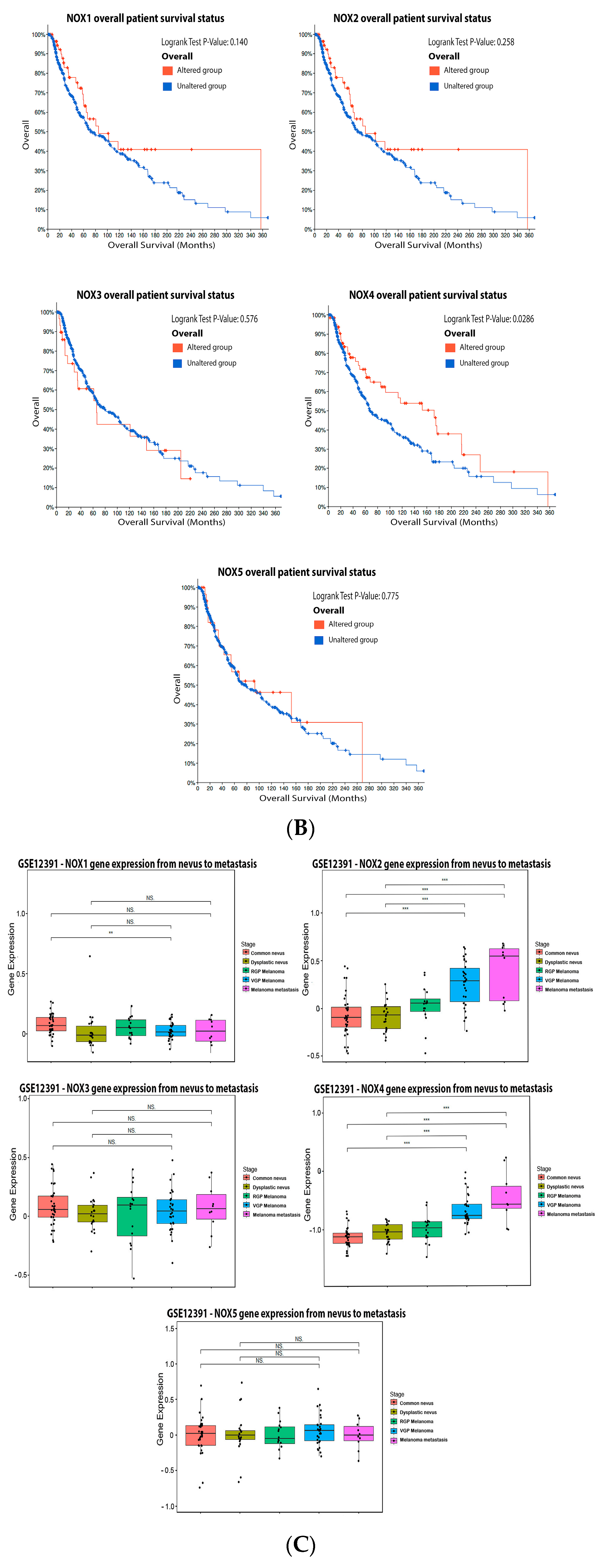
3. RNS and ROS-Detoxifying and Sensitive Enzymes
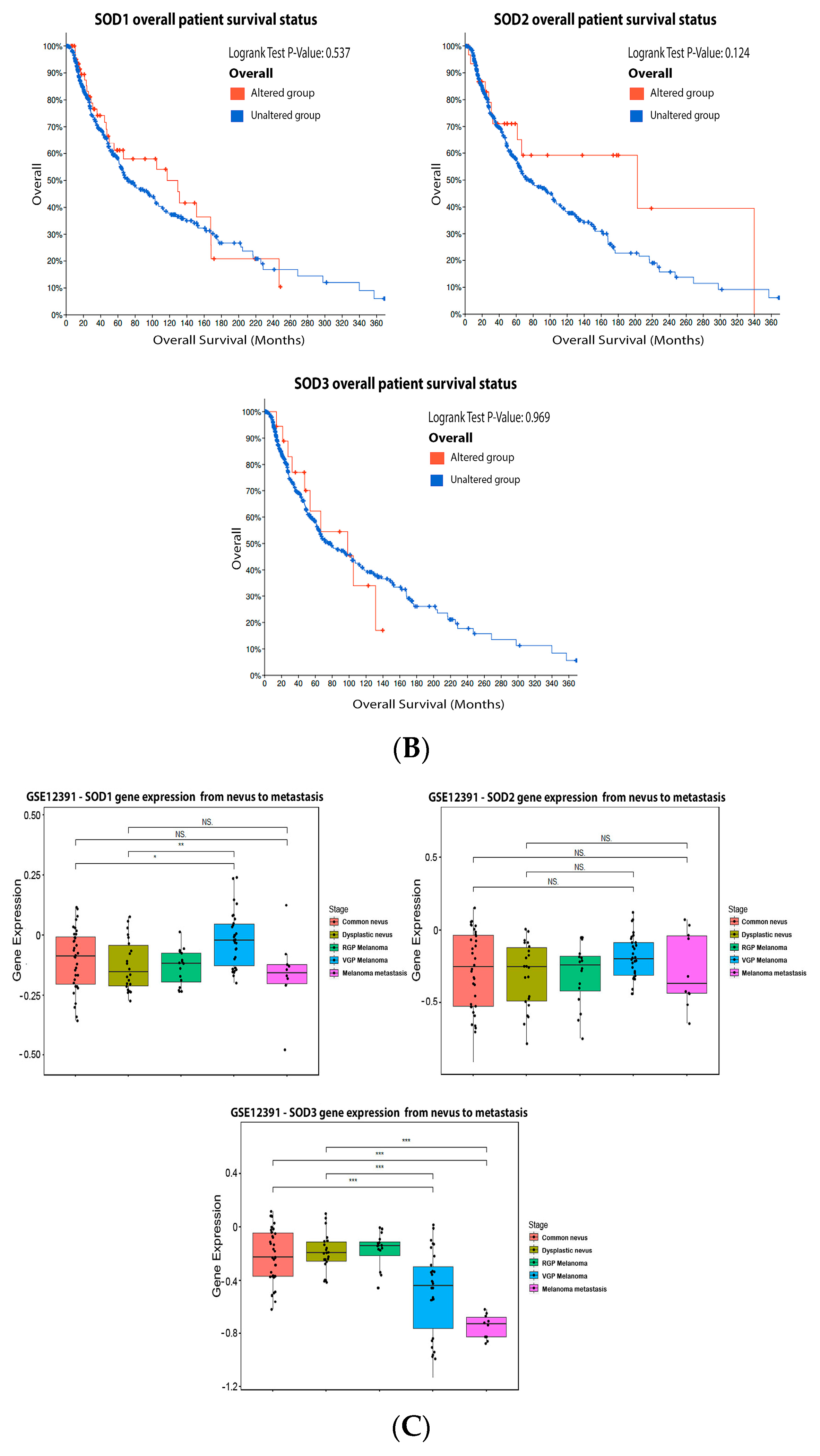
3.1. Superoxide Dismutase (SOD)

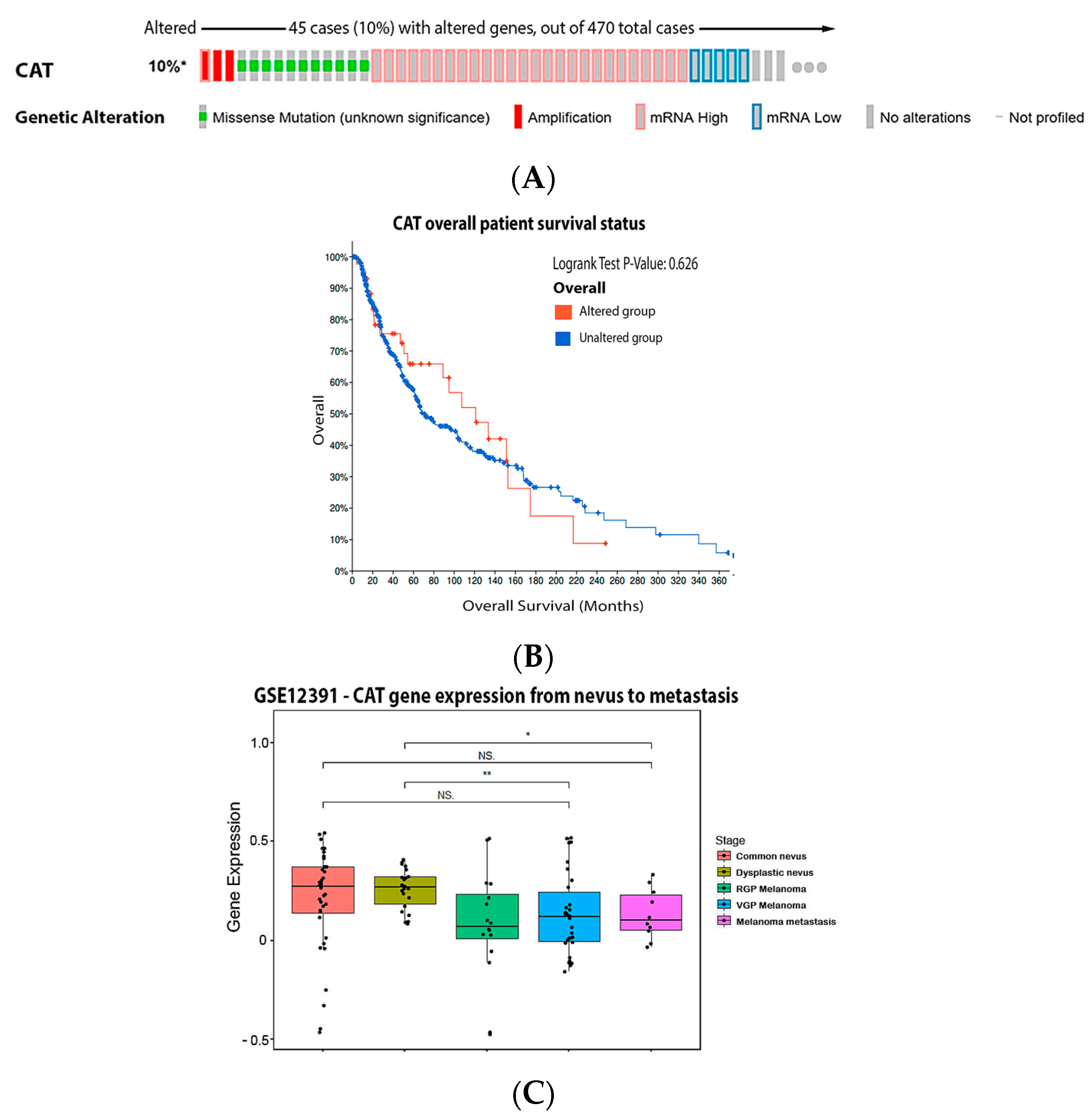
3.2. Catalase (CAT)
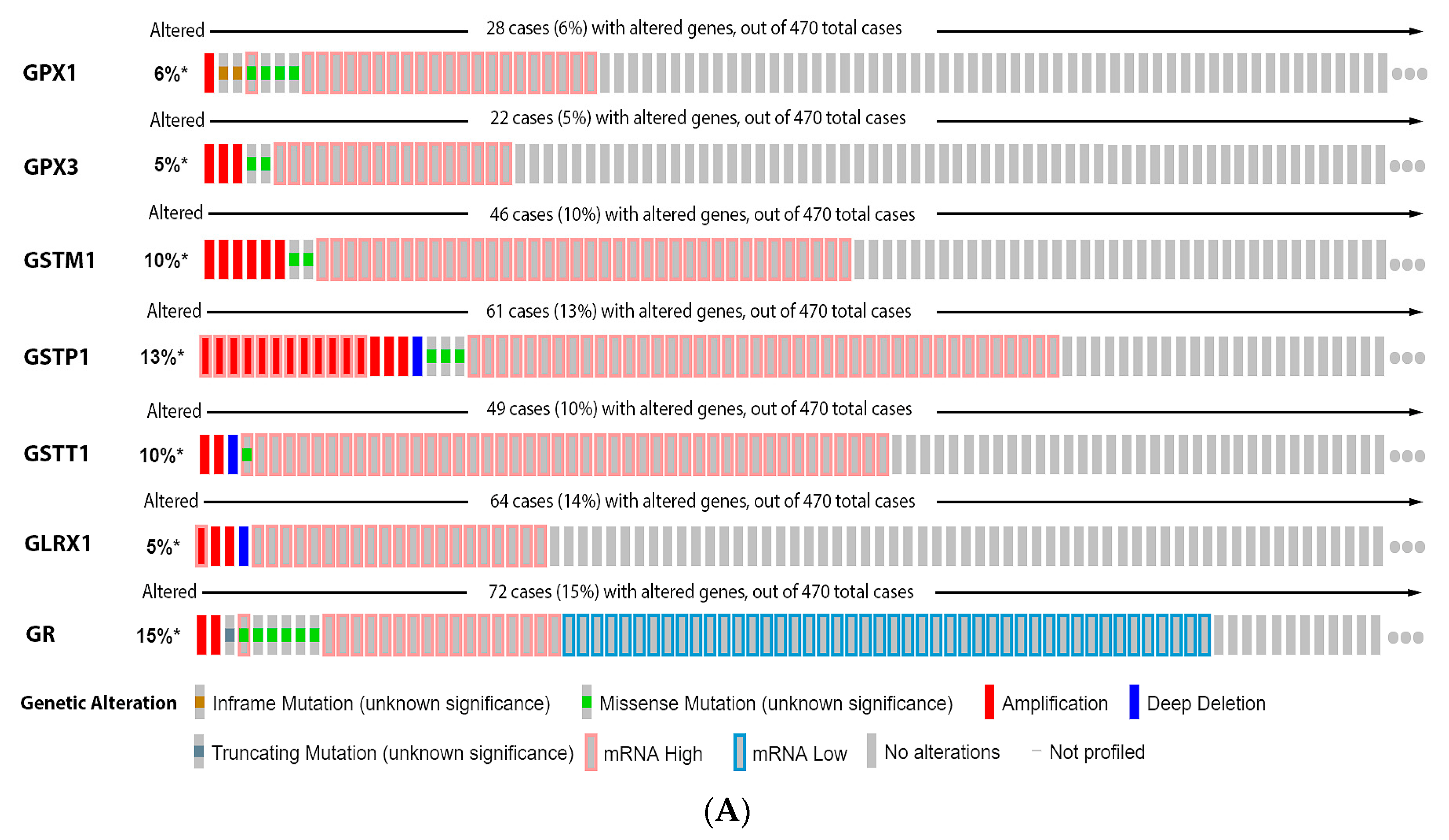
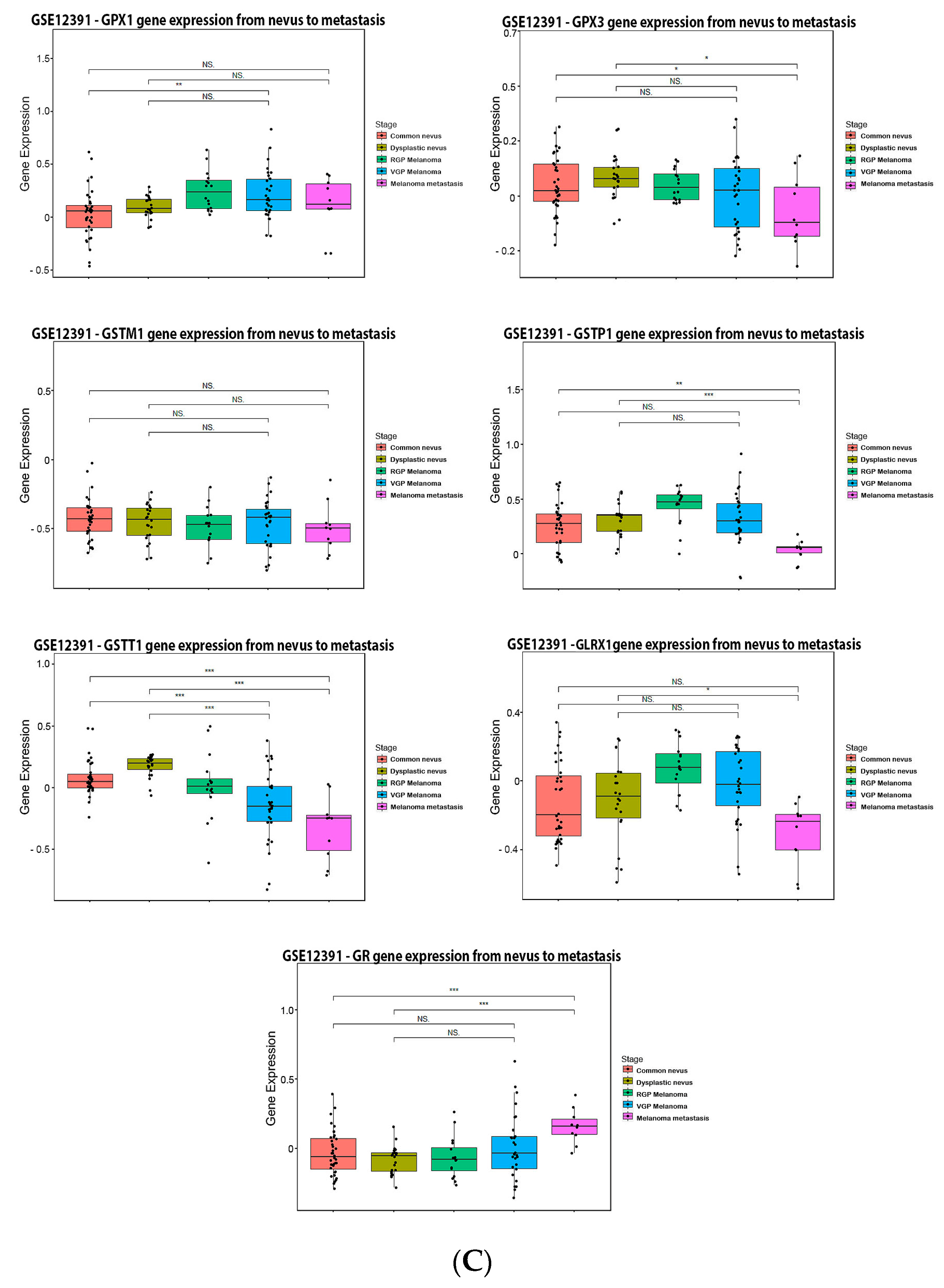
3.3. Glutathione System

3.4. Thioredoxin System

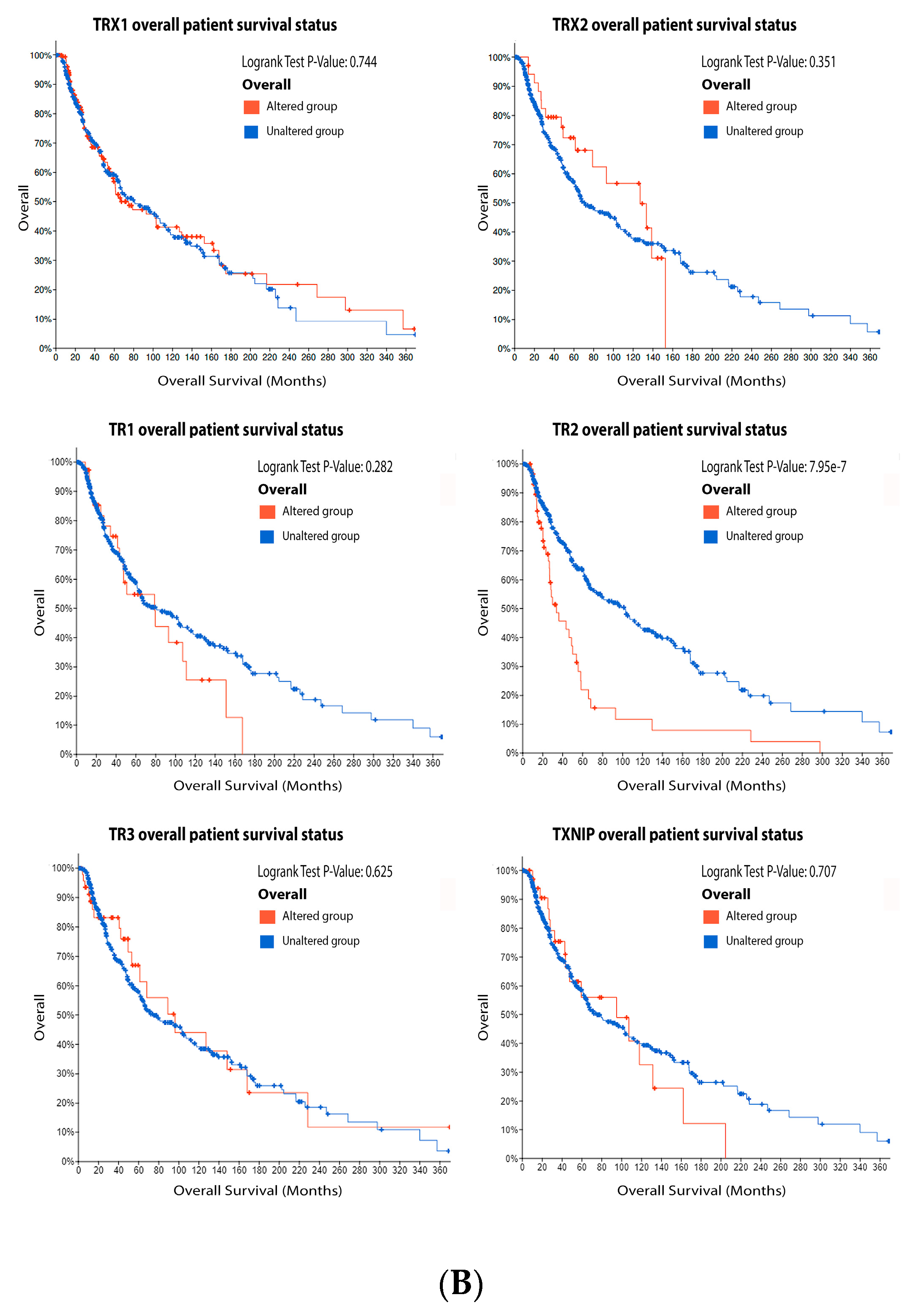
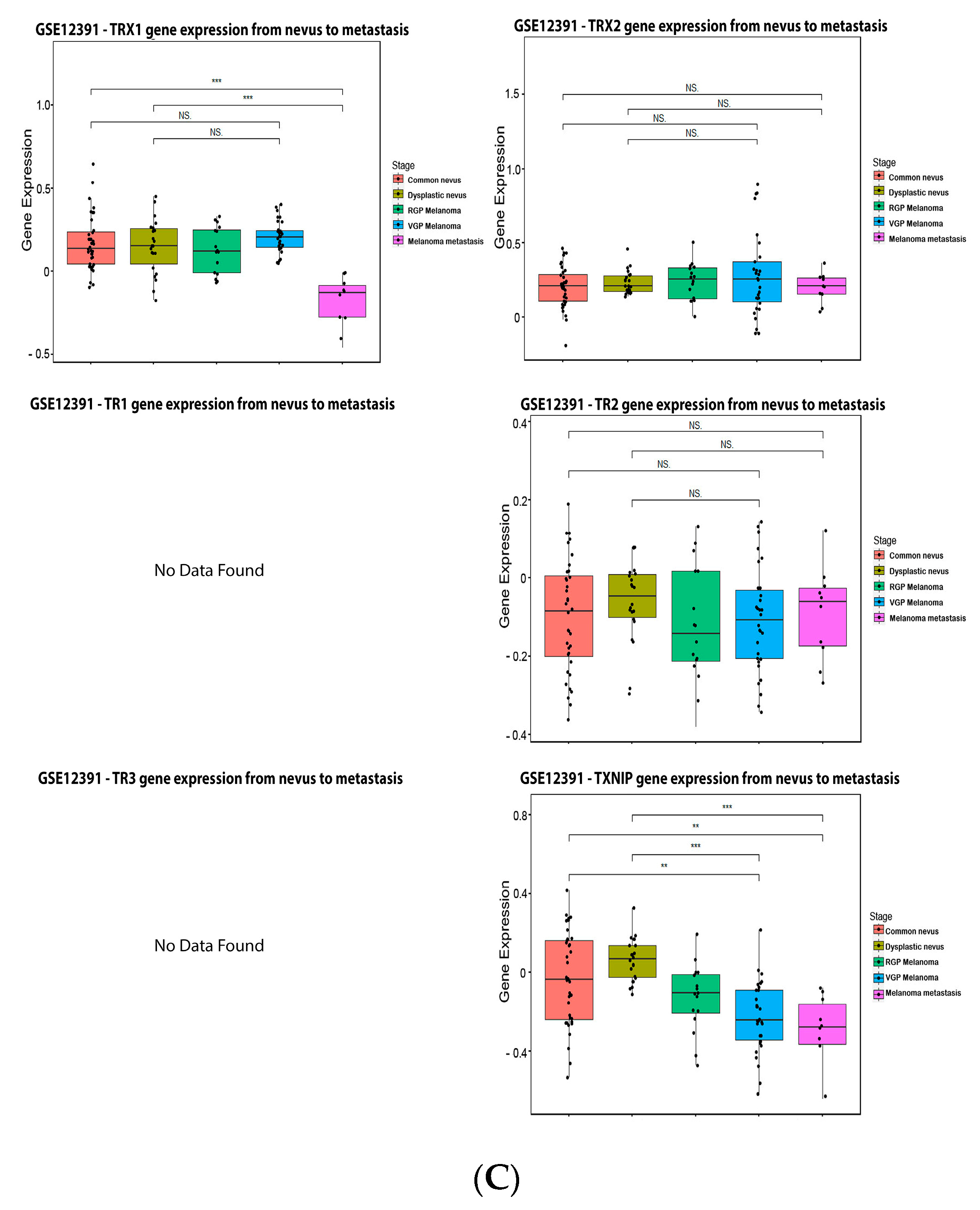 Figure 6. Thioredoxins, thioredoxins reductase and thioredoxin interacting protein on melanoma. (A) Percentage of patients with altered genes and types of genetic alterations; (B) overall patient survival status, and (C) expression in melanoma progression. Red boxes represent the common nevus, yellow boxes the dysplastic nevus, green boxes the radial growth phase (RGP) melanoma, blue boxes the vertical growth phase (VGP) melanoma, and purple boxes the metastatic melanoma. The statistical analysis was performed by ANOVA followed by Tukey test, *** p < 0.001, ** p < 0.01, * p < 0.05 when compared to common or dysplastic nevus. NS: Not significant.
Figure 6. Thioredoxins, thioredoxins reductase and thioredoxin interacting protein on melanoma. (A) Percentage of patients with altered genes and types of genetic alterations; (B) overall patient survival status, and (C) expression in melanoma progression. Red boxes represent the common nevus, yellow boxes the dysplastic nevus, green boxes the radial growth phase (RGP) melanoma, blue boxes the vertical growth phase (VGP) melanoma, and purple boxes the metastatic melanoma. The statistical analysis was performed by ANOVA followed by Tukey test, *** p < 0.001, ** p < 0.01, * p < 0.05 when compared to common or dysplastic nevus. NS: Not significant.3.5. Peroxiredoxin (PRX)
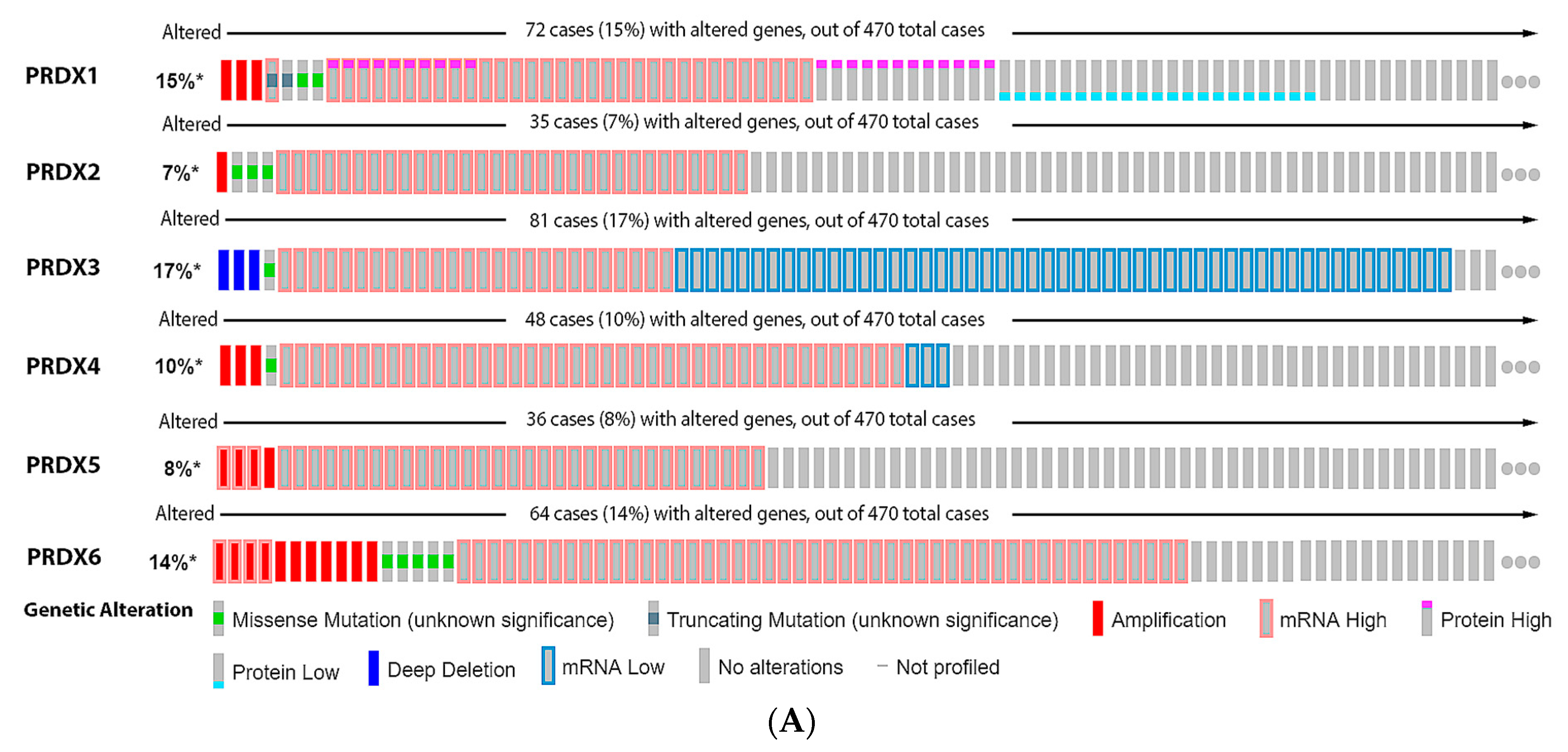

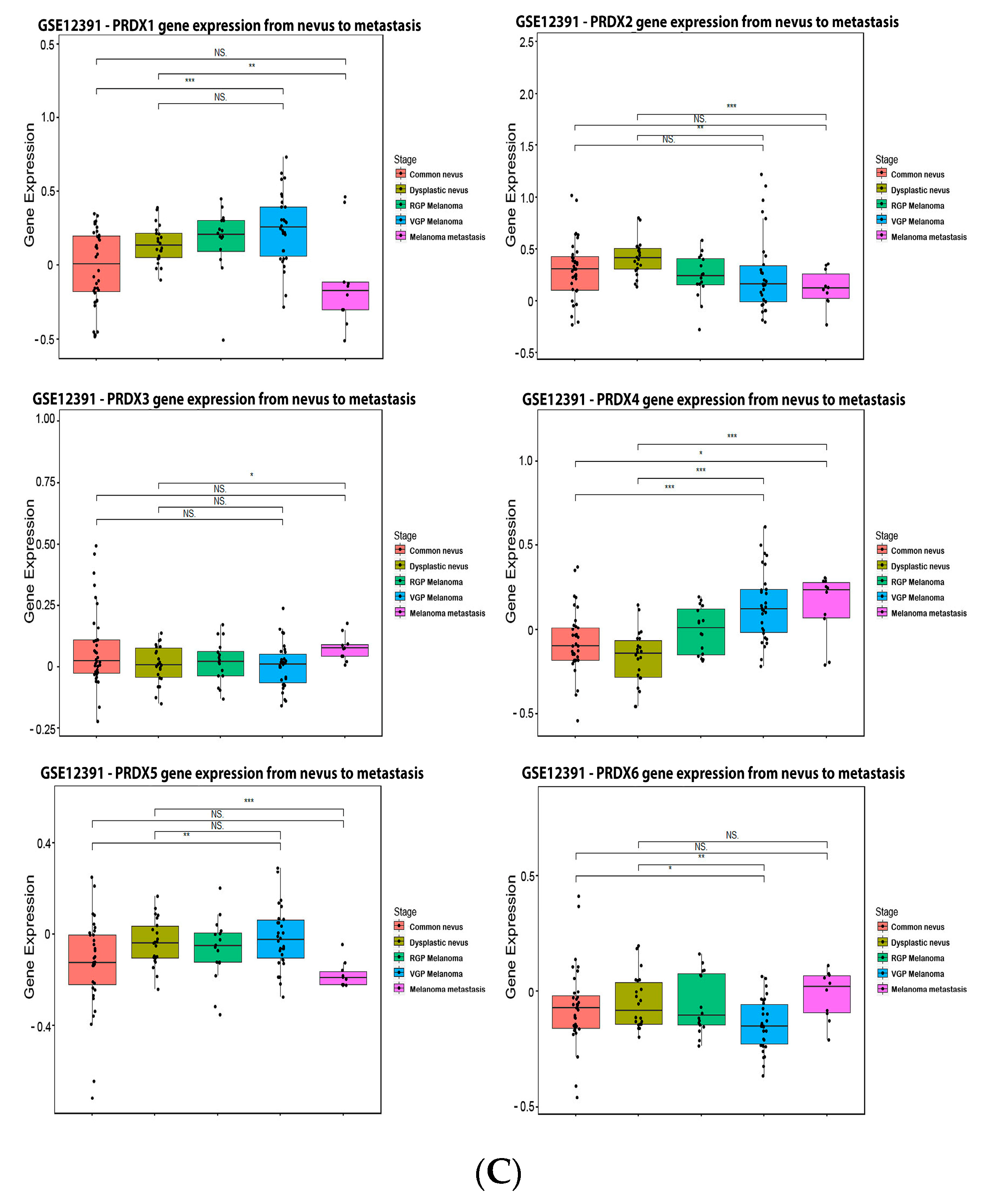
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Cannavò, S.P.; Tonacci, A.; Bertino, L.; Casciaro, M.; Borgia, F.; Gangemi, S. The role of oxidative stress in the biology of melanoma: A systematic review. Pathol. Res. Pract. 2019, 215, 21–28.
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.H.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998.
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65.
- Wittgen, H.G.M.; van Kempen, L.C.L.T. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007, 17, 400–409.
- Purohit, V.; Simeone, D.M.; Lyssiotis, C.A. Metabolic regulation of redox balance in cancer. Cancers 2019, 11, 955.
- Ferraz, L.S.; da Costa, R.T.; da Costa, C.A.; Ribeiro, C.A.J.; Arruda, D.C.; Maria-Engler, S.S.; Rodrigues, T. Targeting mitochondria in melanoma: Interplay between MAPK signaling pathway and mitochondrial dynamics. Biochem. Pharmacol. 2020, 178, 114104.
- Arslanbaeva, L.R.; Santoro, M.M. Adaptive redox homeostasis in cutaneous melanoma. Redox Biol. 2020, 37, 101753.
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315.
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325.
- Yuan, L.; Mishra, R.; Patel, H.; Abdulsalam, S.; Greis, K.D.; Kadekaro, A.L.; Merino, E.J.; Garrett, J.T. Utilization of reactive oxygen species targeted therapy to prolong the efficacy of BRAF inhibitors in melanoma. J. Cancer 2018, 9, 4665–4676.
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 2018, 33, 890–904.e5.
- Bishal Paudel, B.; Lewis, J.E.; Hardeman, K.N.; Hayford, C.E.; Robbins, C.J.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A.; Kemp, M.L.; Quaranta, V. Disruption of redox balance enhances the effects of BRAF-inhibition in melanoma cells. bioRxiv 2019, 818989.
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164.
- Ekshyyan, O.; Aw, T.Y. Decreased susceptibility of differentiated PC12 cells to oxidative challenge: Relationship to cellular redox and expression of apoptotic protease activator factor-1. Cell Death Differ. 2005, 12, 1066–1077.
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55.
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612.
- Brohem, C.A.; Sawada, T.C.H.; Massaro, R.R.; Almeida, R.L.; Rivelli, D.P.; Ropke, C.D.; da Silva, V.V.; de Lima, T.M.; Curi, R.; Barros, S.B.M.; et al. Apoptosis induction by 4-nerolidylcatechol in melanoma cell lines. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2009, 23, 111–119.
- Alves-Fernandes, D.K.; de Oliveira, É.A.; Faião-Flores, F.; Alicea-Rebecca, G.; Weeraratna, A.T.; Smalley, K.S.M.; de Barros, S.B.M.; Maria-Engler, S.S. ER stress promotes antitumor effects in BRAFi/MEKi resistant human melanoma induced by natural compound 4-nerolidylcathecol (4-NC). Pharmacol. Res. 2019, 141, 63–72.
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Alcácer, J.; Benlloch, M.; Pellicer, J.A.; Estrela, J.M. Melanoma in the liver: Oxidative stress and the mechanisms of metastatic cell survival. Semin. Cancer Biol. 2021, 71, 109–121.
- Scatolini, M.; Grand, M.M.; Grosso, E.; Venesio, T.; Pisacane, A.; Balsamo, A.; Sirovich, R.; Risio, M.; Chiorino, G. Altered molecular pathways in melanocytic lesions. Int. J. Cancer 2010, 126, 1869–1881.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the CBioPortal. Sci. Signal. 2013, 6, pl1.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 500–509.
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178.
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 3051–3064.
- Bruch-Gerharz, D.; Ruzicka, T.; Kolb-Bachofen, V. Nitric oxide and its implications in skin homeostasis and disease—A review. Arch. Dermatol. Res. 1998, 290, 643–651.
- Massi, D.; Marconi, C.; Franchi, A.; Bianchini, F.; Paglierani, M.; Ketabchi, S.; Miracco, C.; Santucci, M.; Calorini, L. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: Evidence from human and experimental tumors. Hum. Pathol. 2007, 38, 1516–1525.
- Joshi, M.; Strandhoy, J.; White, W.L. Nitric oxide synthase activity is up-regulated in melanoma cell lines. Melanoma Res. 1996, 6, 121–126.
- Ahmed, B.; van den Oord, J.J. Expression of the inducible isoform of nitric oxide synthase in pigment cell lesions of the skin. Br. J. Dermatol. 2000, 142, 432–440.
- Grimm, E.A.; Ellerhorst, J.; Tang, C.H.; Ekmekcioglu, S. Constitutive intracellular production of INOS and NO in Human melanoma: Possible role in regulation of growth and resistance to apoptosis. Nitric Oxide Biol. Chem. 2008, 19, 133–137.
- Johansson, C.C.; Egyházi, S.; Masucci, G.; Harlin, H.; Mougiakakos, D.; Poschke, I.; Nilsson, B.; Garberg, L.; Tuominen, R.; Linden, D.; et al. Prognostic significance of tumor INOS and COX-2 in stage III malignant cutaneous melanoma. Cancer Immunol. Immunother. 2009, 58, 1085–1094.
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of INOS in cancer. Redox Biol. 2015, 6, 334–343.
- Ekmekcioglu, S.; Ellerhorst, J.; Smid, C.M.; Prieto, V.G.; Munsell, M.; Buzaid, A.C.; Grimm, E.A. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 4768–4775.
- Van den Oord, A. Expression of the neuronal isoform of nitric oxide synthase (NNOS) and its inhibitor, protein inhibitor of NNOS, in pigment cell lesions of the skin. Br. J. Dermatol. 1999, 141, 12–19.
- Huang, H.; Li, H.; Yang, S.; Chreifi, G.; Martásek, P.; Roman, L.J.; Meyskens, F.L.; Poulos, T.L.; Silverman, R.B. Potent and selective double-headed-2-carboximidamide of neuronal nitric oxide synthase for the treatment of melanoma. J. Med. Chem. 2014, 57, 686.
- Yang, Z.; Misner, B.; Ji, H.; Poulos, T.L.; Silverman, R.B.; Meyskens, F.L.; Yang, S. Targeting nitric oxide signaling with NNOS inhibitors as a novel strategy for the therapy and prevention of human melanoma. Antioxid. Redox Signal. 2013, 19, 433–447.
- Weiss, L. Metastatic inefficiency. Adv. Cancer Res. 1990, 54, 159–211.
- Sikora, A.G.; Gelbard, A.; Davies, M.A.; Sano, D.; Ekmekcioglu, S.; Kwon, J.; Hailemichael, Y.; Jayaraman, P.; Myers, J.N.; Grimm, E.A.; et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin. Cancer Res. 2010, 16, 1834–1844.
- Huang, S.-H.; Hsu, M.-H.; Hsu, S.-C.; Yang, J.-S.; Huang, W.-W.; Huang, A.-C.; Hsiao, Y.-P.; Yu, C.-C.; Chung, J.-G. Phenethyl isothiocyanate triggers apoptosis in human malignant melanoma A375.S2 cells through reactive oxygen species and the mitochondria-dependent pathways. Hum. Exp. Toxicol. 2014, 33, 270–283.
- Huang, Y.; Yu, P.; Li, W.; Ren, G.; Roberts, A.I.; Cao, W.; Zhang, X.; Su, J.; Chen, X.; Chen, Q.; et al. P53 regulates mesenchymal stem cell-mediated tumor suppression in a tumor microenvironment through immune modulation. Oncogene 2014, 33, 3830–3838.
- Place, A.E.; Suh, N.; Williams, C.R.; Risingsong, R.; Honda, T.; Honda, Y.; Gribble, G.W.; Leesnitzer, L.M.; Stimmel, J.B.; Willson, T.M.; et al. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 2798–2806.
- Ding, Z.; Qin, Y.; Kim, S.-H.; Grimm, E.A. Nitric oxide activates the PI3Kinase-akt pathway in human melanoma cells. Free Radic. Biol. Med. 2016, 100, S120.
- Yang, J.; Wu, L.J.; Tashiro, S.I.; Onodera, S.; Ikejima, T. Nitric oxide activated by P38 and NF-KappaB facilitates apoptosis and cell cycle arrest under oxidative stress in evodiamine-treated human melanoma A375-S2 cells. Free Radic. Res. 2008, 42, 1–11.
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272.
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875.
- Juhasz, A.; Ge, Y.; Markel, S.; Chiu, A.; Matsumoto, L.; van Balgooy, J.; Roy, K.; Doroshow, J.H. Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic. Res. 2009, 43, 523–532.
- Liu, F.; Gomez Garcia, A.M.; Meyskens, F.L. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial–mesenchymal transition in melanoma cells. J. Investig. Dermatol. 2012, 132, 2033–2041.
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654.
- Ribeiro-Pereira, C.; Moraes, J.A.; de Jesus Souza, M.; Laurindo, F.R.; Arruda, M.A.; Barja-Fidalgo, C. Redox Modulation of FAK Controls melanoma survival—Role of NOX4. PLoS ONE 2014, 9, e99481.
- Antony, S.; Jiang, G.; Wu, Y.; Meitzler, J.L.; Makhlouf, H.R.; Haines, D.C.; Butcher, D.; Hoon, D.S.; Ji, J.; Zhang, Y.; et al. NADPH oxidase 5 (NOX5)-induced reactive oxygen signaling modulates normoxic HIF-1α and P27 Kip1 expression in malignant melanoma and other human tumors. Mol. Carcinog. 2017, 56, 2643–2662.
- Beyerstedt, S.; Franco, M.; Oliveira, T.; Mendonça, G.; Alves-Fernandes, D.; Maria-Engler, S.; Machado-Neto, J.; Lopes, L. Targeting protein disulfide isomerase to overcome resistance to BRAF inhibitors in melanoma. Free Radic. Biol. Med. 2018, 128, S62.
- Prasad, R.; Kappes, J.C.; Katiyar, S.K. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget 2016, 7, 7899–7912.
- Zhao, Y.; Liu, J.; McMartin, K.E. Inhibition of NADPH oxidase activity promotes differentiation of B16 melanoma cells. Oncol. Rep. 2008, 19, 1225–1230.
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349.
- Kwee, J.K.; Mitidieri, E.; Affonso, O.R. Lowered superoxide dismutase in highly metastatic B16 melanoma cells. Cancer Lett. 1991, 57, 199–202.
- Church, S.L.; Grant, J.W.; Ridnour, L.A.; Oberley, L.W.; Swanson, P.E.; Meltzer, P.S.; Trent, J.M. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc. Natl. Acad. Sci. USA 1993, 90, 3113–3117.
- Schadendorf, D.; Zuberbier, T.; Diehl, S.; Schadendorf, C.; Czarnetzki, B.M. Serum manganese superoxide dismutase is a new tumour marker for malignant melanoma. Melanoma Res. 1995, 5, 351–353.
- Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF mutant melanoma adjusts to BRAF/MEK inhibitors via dependence on increased antioxidant SOD2 and increased reactive oxygen species levels. Cancers 2020, 12, 1661.
- Radojičić, R.; Spasić, M.; Simić, J.; Petrović, V.M.I. Effect of bovine Cu, Zn superoxide dismutase on C3 Clone of B-16 mouse melanoma cells in the culture. J. Steroid Biochem. 1987, 28, 118.
- Radojicic, R.; Spasic, S.; Saicic, Z.; Jovanovic, T.; Simic-Krstic, J. Superoxide dismutase activity as a function of culture aging of B-16 mouse melanoma cells. J. Serb. Chem. Soc. 2004, 69, 1005–1011.
- Wheeler, M.D.; Smutney, O.M.; Samulski, R.J. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of b16 melanomas in mice. Mol. Cancer Res. MCR 2003, 1, 871–881.
- Robbins, D.; Zhao, Y. The role of manganese superoxide dismutase in skin cancer. Enzym. Res. 2011, 2011, 409295.
- Grammatico, P.; Maresca, V.; Roccella, F.; Roccella, M.; Biondo, L.; Catricalà, C.; Picardo, M. Increased sensitivity to peroxidizing agents is correlated with an imbalance of antioxidants in normal melanocytes from melanoma patients. Exp. Dermatol. 1998, 7, 205–212.
- Jaworska, A.; Stojcevic-Lemic, N.; Nias, A.H.W.; Sies, H. The effect of paraquat on the radiosensitivity of melanoma cells: The role of superoxide dismutase & CATALASE. Free Radic. Res. 2009, 18, 139–145.
- Bisevac, J.P.; Djukic, M.; Stanojevic, I.; Stevanovic, I.; Mijuskovic, Z.; Djuric, A.; Gobeljic, B.; Banovic, T.; Vojvodic, D. Association between oxidative stress and melanoma progression. J. Med. Biochem. 2018, 37, 12–20.
- Bracalente, C.; Ibañez, I.L.; Berenstein, A.; Notcovich, C.; Cerda, M.B.; Klamt, F.; Chernomoretz, A.; Durán, H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Upregulation of Antioxidant genes correlates with regression of melanoma malignancy and with malignant progression when downregulated. Oncotarget 2016, 7, 41154–41171.
- Hyoudou, K.; Nishikawa, M.; Kobayashi, Y.; Umeyama, Y.; Yamashita, F.; Hashida, M. PEGylated catalase prevents metastatic tumor growth aggravated by tumor removal. Free Radic. Biol. Med. 2006, 41, 1449–1458.
- Hyoudou, K.; Nishikawa, M.; Umeyama, Y.; Kobayashi, Y.; Yamashita, F.; Hashida, M. Inhibition of metastatic tumor growth in mouse lung by repeated administration of polyethylene glycol-conjugated catalase: Quantitative analysis with firefly luciferase-expressing melanoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7685–7691.
- Meyskens, F.L.; Farmer, P.; Fruehauf, J.P. Redox regulation in human melanocytes and melanoma. Pigment Cell Res. 2001, 14, 148–154.
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8.
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis 1999, 17, 567–574.
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191.
- Ortega, A.L.; Carretero, J.; Obrador, E.; Gambini, J.; Asensi, M.; Rodilla, V.; Estrela, J.M. Tumor cytotoxicity by endothelial cells impairment of the mitochondrial system for glutathione uptake in mouse B16 melanoma cells that survive after in vitro interaction with the hepatic sinusoidal endothelium. J. Biol. Chem. 2003, 278, 13888–13897.
- Woźniak, A.; Drewa, G.; Woźniak, B.; Schachtschabel, D.O. Activity of antioxidant enzymes and concentration of lipid peroxidation products in selected tissues of mice of different ages, both healthy and melanoma-bearing. Z. Gerontol. Geriatr. 2004, 37, 184–189.
- Bansal, A.; Celeste Simon, M. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298.
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666.
- Conticello, C.; Martinetti, D.; Adamo, L.; Buccheri, S.; Giuffrida, R.; Parrinello, N.; Lombardo, L.; Anastasi, G.; Amato, G.; Cavalli, M.; et al. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer 2012, 131, 2197–2203.
- O’dwyer, P.J.; Hamilton, T.C.; Young, R.C.; Lacreta, F.P.; Carp, N.; Tew, K.D.; Padavic, K.; Comis, R.L.; Ozols, R.F. Depletion of glutathione in normal and malignant human cells in vivo by buthionine sulfoximine: Clinical and biochemical results. J. Natl. Cancer Inst. 1992, 84, 264–267.
- Bailey, H.H.; Mulcahy, R.T.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Tombes, M.B.; Wilding, G.; Pomplun, M.; Spriggs, D.R. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: An attempt at modulation of glutathione. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1994, 12, 194–205.
- Fruehauf, J.P.; Zonis, S.; Al-Bassam, M.; Kyshtoobayeva, A.; Dasgupta, C.; Milovanovic, T.; Parker, R.J.; Buzaid, A.C. Melanin content and downregulation of glutathione S-transferase contribute to the action of L-buthionine-S-sulfoximine on human melanoma. Chem. Biol. Interact. 1998, 111-112, 277–305.
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092.
- Beberok, A.; Wrześniok, D.; Szlachta, M.; Rok, J.; Rzepka, Z.; Respondek, M.; Buszman, E. Lomefloxacin induces oxidative stress and apoptosis in COLO829 melanoma cells. Int. J. Mol. Sci. 2017, 18, 2194.
- Schott, M.; de Jel, M.M.; Engelmann, J.C.; Renner, P.; Geissler, E.K.; Bosserhoff, A.K.; Kuphal, S. Selenium-binding protein 1 is down-regulated in malignant melanoma. Oncotarget 2018, 9, 10445.
- Chen, H.; Zheng, Z.; Kim, K.Y.; Jin, X.; Roh, M.R.; Jin, Z. Hypermethylation and downregulation of glutathione peroxidase 3 are related to pathogenesis of melanoma. Oncol. Rep. 2016, 36, 2737–2744.
- Yi, Z.; Jiang, L.; Zhao, L.; Zhou, M.; Ni, Y.; Yang, Y.; Yang, H.; Yang, L.; Zhang, Q.; Kuang, Y.; et al. glutathione peroxidase 3 (GPX3) suppresses the growth of melanoma cells through reactive oxygen species (ROS)-dependent stabilization of hypoxia-inducible factor 1-α and 2-α. J. Cell. Biochem. 2019, 120, 19124–19136.
- Schadendorf, D.; Jurgovsky, K.; Kohlmus, C.M.; Czarnetzki, B.M. Glutathione and related enzymes in tumor progression and metastases of human melanoma. J. Investig. Dermatol. 1995, 105, 109–112.
- Nogués, M.R.; Giralt, M.; Cervelló, I.; del Castillo, D.; Espeso, O.; Argany, N.; Aliaga, A.; Mallol, J. Parameters related to oxygen free radicals in human skin: A study comparing healthy epidermis and skin cancer tissue. J. Investig. Dermatol. 2002, 119, 645–652.
- Brigelius-Flohé, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta 2009, 1790, 1555–1568.
- Ji, Y.; Dai, F.; Yan, S.; Shi, J.Y.; Zhou, B. Identification of catechol-type diphenylbutadiene as a tyrosinase-activated pro-oxidative chemosensitizer against melanoma A375 cells via glutathione S-transferase inhibition. J. Agric. Food Chem. 2019, 67, 9060–9069.
- Kanetsky, P.A.; Holmes, R.; Walker, A.; Najarian, D.; Swoyer, J.; Guerry, D.; Halpern, A.; Rebbeck, T.R. Interaction of glutathione S-transferase M1 and T1 genotypes and malignant melanoma. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2001, 10, 509–513.
- Yura, Y.; Johnson, R.; Watanabe, Y.; Tsukahara, Y.; Ferran, B.P.; Murdoch, C.; van der Velden, J.; Bachschmid, M.M.; Heininger, Y.J.; Matsui, R. Differential regulation of ischemic limb vascularization and tumor growth by endothelial glutaredoxin-1. Free Radic. Biol. Med. 2017, 112, 38–39.
- Schallreuter, K.U.; Wood, J.M. Sensitivity and resistance in human metastatic melanoma to the new chloroethylnitrosourea anti-tumor drug fotemustine. Biochim. Biophys. Acta 1991, 1096, 277–283.
- Schallreuter, K.U.; Gleason, F.K.; Wood, J.M. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase and ribonucleotide reductase. Biochim. Biophys. Acta 1990, 1054, 14–20.
- Fruehauf, J.P. BCNU-Mediated Glutathione Depletion and Inhibition of Glutathione Reductase, Ribonucleotide Reductase, and DNA Synthesis: Novel Mechanisms of Antineoplastic Activity. Ph.D. Thesis, Rush University, College of Nursing, Chicago, IL, USA, 1996.
- Chong, B.S.H. The Role of Glutaredoxin-1 on B16F0 Melanoma Growth and Angiogenesis in Diet-Induced Diabetic Mice. Master’s Thesis, Boston University, School of Medicine, Boston, MA, USA, 2015.
- Li, X.; Wu, J.; Zhang, X.; Chen, W. Glutathione reductase-mediated thiol oxidative stress suppresses metastasis of murine melanoma cells. Free Radic. Biol. Med. 2018, 129, 256–267.
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87.
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470.
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433.
- Chakraborty, P.; Chatterjee, S.; Kesarwani, P.; Thyagarajan, K.; Iamsawat, S.; Dalheim, A.; Nguyen, H.; Selvam, S.P.; Nasarre, P.; Scurti, G.; et al. Thioredoxin-1 improves the immunometabolic phenotype of antitumor T cells. J. Biol. Chem. 2019, 294, 9198–9212.
- Wang, X.; Dong, H.; Li, Q.; Li, Y.; Hong, A. Thioredoxin induces tregs to generate an immunotolerant tumor microenvironment in metastatic melanoma. Oncoimmunology 2015, 4, e1027471.
- Cheng, G.C.; Schulze, P.C.; Lee, R.T.; Sylvan, J.; Zetter, B.R.; Huang, H. Oxidative stress and thioredoxin-interacting protein promote intravasation of melanoma cells. Exp. Cell Res. 2004, 300, 297–307.
- Song, H.; Cho, D.; Jeon, J.H.; Han, S.H.; Hur, D.Y.; Kim, Y.S.; Choi, I. Vitamin D3 up-regulating protein 1 (VDUP1) antisense DNA regulates tumorigenicity and melanogenesis of murine melanoma cells via regulating the expression of fas ligand and reactive oxygen species. Immunol. Lett. 2003, 86, 235–247.
- Goldberg, S.F.; Miele, M.E.; Hatta, N.; Takata, M.; Paquette-Straub, C.; Freedman, L.P.; Welch, D.R. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–440.
- Li, K.; Tang, M.; Tong, S.; Wang, C.; Sun, Q.; Lv, M.; Sun, X.; Wang, T.; Jin, S. BRAFi induced demethylation of MiR-152-5p regulates phenotype switching by targeting TXNIP in cutaneous melanoma. Apoptosis 2020, 25, 179–191.
- Cassidy, P.B.; Honeggar, M.; Poerschke, R.L.; White, K.; Florell, S.R.; Andtbacka, R.H.I.; Tross, J.; Anderson, M.; Leachman, S.A.; Moos, P.J. The role of thioredoxin reductase 1 in melanoma metabolism and metastasis. Pigment Cell Melanoma Res. 2015, 28, 685–695.
- Cassidy, P.; Kline, C.; Carpenter, E.; Laws, M.; Moos, P.; Indra, A.; Leachman, S. Thioredoxin reductase 1 knockdown disrupts pigment synthesis in melanocytes. Free Radic. Biol. Med. 2018, 128, S64.
- Zheng, X.; Chen, Y.; Bai, M.; Liu, Y.; Xu, B.; Sun, R.; Zeng, H. The antimetastatic effect and underlying mechanisms of thioredoxin reductase inhibitor ethaselen. Free Radic. Biol. Med. 2019, 131, 7–17.
- Sachweh, M.C.C.; Stafford, W.C.; Drummond, C.J.; McCarthy, A.R.; Higgins, M.; Campbell, J.; Brodin, B.; Arnér, E.S.J.; Laín, S. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget 2015, 6, 16488–16506.
- Cao, Z.; Lindsay, J.G.; Isaacs, N.W. Mitochondrial peroxiredoxins. Sub Cell. Biochem. 2007, 44, 295–315.
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325.
- Carvalho, L.A.C.; Truzzi, D.R.; Fallani, T.S.; Alves, S.V.; Toledo, J.C.; Augusto, O.; Netto, L.E.S.; Meotti, F.C. Urate hydroperoxide oxidizes human peroxiredoxin 1 and peroxiredoxin 2. J. Biol. Chem. 2017, 292, 8705–8715.
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561.
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin involvement in the initiation and progression of human cancer. Antioxid. Redox Signal. 2018, 28, 591–608.
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653.
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40.
- Rhee, S.G. Overview on peroxiredoxin. Mol. Cells 2016, 39, 1–5.
- Peskin, A.V.; Pace, P.E.; Behring, J.B.; Paton, L.N.; Soethoudt, M.; Bachschmid, M.M.; Winterbourn, C.C. Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J. Biol. Chem. 2016, 291, 3053–3062.
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530.
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.D.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70.
- Hintsala, H.R.; Soini, Y.; Haapasaari, K.M.; Karihtala, P. Dysregulation of redox-state-regulating enzymes in melanocytic skin tumours and the surrounding microenvironment. Histopathology 2015, 67, 348–357.
- Lee, D.J.; Kang, D.H.; Choi, M.; Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, Y.J.; Lee, K.W.; Kang, S.W. Peroxiredoxin-2 represses melanoma metastasis by increasing E-cadherin/β-catenin complexes in adherens junctions. Cancer Res. 2013, 73, 4744–4757.
- Furuta, J.; Nobeyama, Y.; Umebayashi, Y.; Otsuka, F.; Kikuchi, K.; Ushijima, T. Silencing of peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006, 66, 6080–6086.
- Chung, Y.M.; Yoo, Y.D.; Park, J.K.; Kim, Y.T.; Kim, H.J. Increased expression of peroxiredoxin II confers resistance to cisplatin. Anticancer Res. 2001, 21, 1129–1133.
- Sharapov, M.G.; Novoselov, V.I. Catalytic and signaling role of peroxiredoxins in carcinogenesis. Biochem. Biokhimiia 2019, 84, 79–100.
- Schmitt, A.; Schmitz, W.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Peroxiredoxin 6 triggers melanoma cell growth by increasing arachidonic acid-dependent lipid signalling. Biochem. J. 2015, 471, 267–279.
- Stone, W.L.; Krishnan, K.; Palau, V.E.; Lightner, J.W.; Brannon, M.F. AKT1 activation up regulates peroxiredoxin 1 in human melanoma cells. Free Radic. Biol. Med. 2016, 100, S130.
- Zykova, T.A.; Zhu, F.; Vakorina, T.I.; Zhang, J.; Higgins, L.A.; Urusova, D.V.; Bode, A.M.; Dong, Z. T-LAK cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of prx1 peroxidase activity. J. Biol. Chem. 2010, 285, 29138–29146.
- Sinha, P.; Poland, J.; Kohl, S.; Schnölzer, M.; Helmbach, H.; Hütter, G.; Lage, H.; Schadendorf, D. Study of the development of chemoresistance in melanoma cell lines using proteome analysis. Electrophoresis 2003, 24, 2386–2404.
- Nawarak, J.; Huang-Liu, R.; Kao, S.H.; Liao, H.H.; Sinchaikul, S.; Chen, S.T.; Cheng, S.L. Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim. Biophys. Acta 2009, 1794, 159–167.
- Ramasamy, P.; Ramasamy, P.; Ramasamy, P.; Larkin, A.M.; Larkin, A.M.; Linge, A.; Tiernan, D.; McAree, F.; Horgan, N.; Moriarty, P.; et al. PRDX3 is associated with metastasis and poor survival in uveal melanoma. J. Clin. Pathol. 2020, 73, 408–412.


