Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandru Necula | + 3160 word(s) | 3160 | 2022-02-22 05:08:45 | | | |
| 2 | Peter Tang | Meta information modification | 3160 | 2022-03-04 03:16:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Necula, A. Anatomical-MRI Correlations in Hypertrophic Cardiomyopathy Adults and Children. Encyclopedia. Available online: https://encyclopedia.pub/entry/20169 (accessed on 23 July 2026).
Necula A. Anatomical-MRI Correlations in Hypertrophic Cardiomyopathy Adults and Children. Encyclopedia. Available at: https://encyclopedia.pub/entry/20169. Accessed July 23, 2026.
Necula, Alexandru. "Anatomical-MRI Correlations in Hypertrophic Cardiomyopathy Adults and Children" Encyclopedia, https://encyclopedia.pub/entry/20169 (accessed July 23, 2026).
Necula, A. (2022, March 03). Anatomical-MRI Correlations in Hypertrophic Cardiomyopathy Adults and Children. In Encyclopedia. https://encyclopedia.pub/entry/20169
Necula, Alexandru. "Anatomical-MRI Correlations in Hypertrophic Cardiomyopathy Adults and Children." Encyclopedia. Web. 03 March, 2022.
Copy Citation
Hypertrophic Cardiomyopathy (HCM) is the most frequent hereditary cardiovascular disease and the leading cause of sudden cardiac death in young individuals. Advancements in CMR imaging have allowed for earlier identification and more accurate prognosis of HCM. CMR has been validated as a technique with high sensitivity and specificity, very few contraindications, a low risk of side effects, and is overall a good tool to be employed in the management of HCM patients.
hypertrophic cardiomyopathy
interstitial fibrosis
myocyte disarray
cardiac MRI
delayed enhancement
apical aneurysm
sudden cardiac death
1. Introduction
Hypertrophic cardiomyopathy (HCM) is a primary diffuse or segmental left ventricular hypertrophy in the absence of secondary causes capable of producing hypertrophy. It is the most common congenital cardiac disease, having a prevalence of 1 in 500 people and representing an important cause of sudden death in adolescents [1]. HCM is an autosomal dominant disorder caused by mutations in the 11 sarcomeric genes, encoding heart sarcomere components [2][3]. The beta-myosin heavy chain gene, myosin-binding protein C gene, and troponin T gene are the most frequently mutated genes [4][5]. Genetic testing currently detects a known pathogenic or probable pathogenic mutation in 30–40% of patients with phenotypic HCM [6]. HCM is defined by a maximal wall thickness greater than 15 mm in general population (or greater than 13 mm in patients with a family history of HCM) and by a septal to left posterior wall thickness ratio greater than 1.3 in normotensive patients and greater than 1.5 in hypertensive patients [7][8]. In children, HCM is defined as an increase in LV wall thickness of more than 2 standard deviations from the mean value for age and BMI [9]. In most cases, LV hypertrophy is asymmetric, affecting mostly the interventricular septum, while hypertrophy can occur in a variety of places, including the apex, anterolateral wall, and free wall [10]. Left ventricular outflow tract (LVOT) obstruction, mitral valve apparatus abnormalities such as systolic anterior motion (SAM) or mitral regurgitation (MR), myocardial ischemia, myocardial fibrosis, and disarray are the key characteristics of HCM that explain the pathophysiology of the condition [11] (Figure 1).
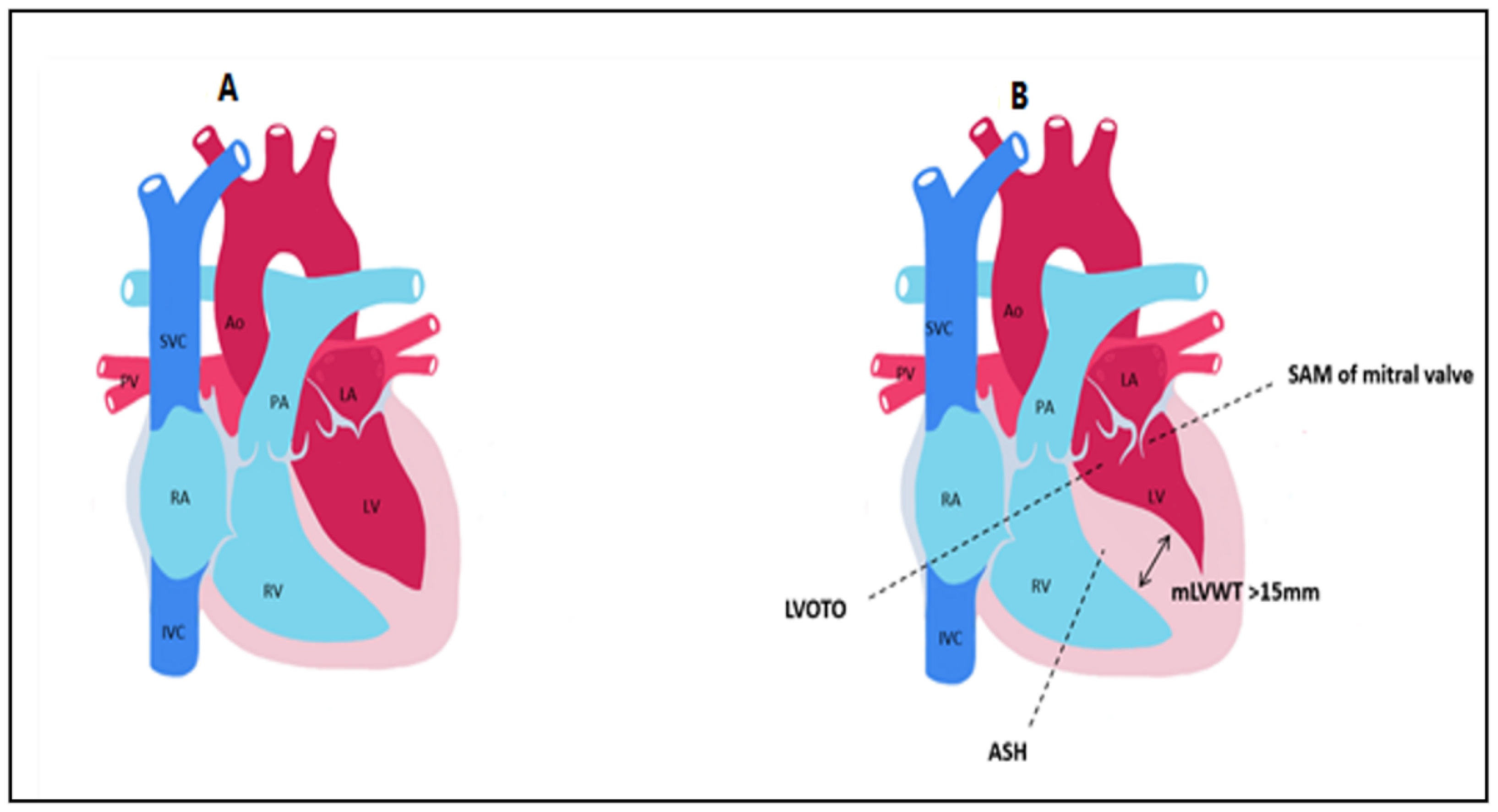
Figure 1. Main anatomical aspects found in hypertrophic cardiomyopathy compared to control. (A) normal heart. (B) HCM LVOTO = Left ventricular outflow tract obstruction; SAM = systolic anterior motion; ASH = asymmetric septal hypertrophy; mLVWT = maximal left ventricular wall thickness; Ao = aorta; PA = pulmonary artery; LA = left atrium; LV = left ventricle; PV = pulmonary veins; RA = right atrium; RV = right ventricle; SVC = superior vena cava; IVC = inferior vena cava.
2. An Anatomical and Histopathological Review of Hypertrophic Cardiomyopathy
The microscopic alterations of the left ventricle in HCM have been recognized since 1957, when Donand Teare investigated [12] the causes of sudden death in eight individuals aged 14 to 44 who had nonspecific symptoms such as dyspnea, fatigue, and mild pectoral angina. On histopathological examination of hypertrophied hearts, the author noticed myocardial disarray, which consisted of an abnormal arrangement of muscle fibers and connective fibrous tissue, clefts and fissures, forming small channels between the two ventricles, preventing the heart from contracting efficiently.
The most prominent diagnostic aspects during a histological examination are: myocyte fiber disarray, interstitial, perivascular, and plexiform fibrosis, as well as small vessel disease, with dysplasia of small intramural coronary arterioles which manifest not only in fibrotic regions but also in areas surrounded by normal myocytes. Myofiber disarray is defined by the torsion of myocyte bundles, and also individual myocytes, with contractile components inside the sarcomere arranged perpendicularly or obliquely. Cardiac cells have larger, pleomorphic, and hyperchromatic nuclei. As a result, heart tissue contractility decreases, and this is compensated by cellular divisions.
In several studies LV hypertrophy was predominantly seen at the level of the interventricular septum and the anterior ventricular wall, also extending to the mitral valves and causing fibrosis. Sutton et al. analyzed the histopathology of 40 necropsy hearts from patients with HCM (10), congestive cardiomyopathy (10), aortic valve stenosis (10) and normal hearts (10) and evaluated the degree of fibrosis and fiber disarray using a four-stage grading system. The findings revealed that fiber disarray was significantly increased in all areas, despite the fact that fibrosis was not severe and the fiber disarray did not correlate with the wall thickness. Patients with hypertrophic cardiomyopathy showed plexiform fibrosis, however, this was not specific for the diagnosis [13]. Later, Maron et al. [14]. observed that myocyte disarray occurred in 88% of HCM patients during autopsy, 70% exhibiting myocardial fibrosis and 56% displaying thickened intramural arterioles with luminal narrowing. Maximal ventricular septal thickness ranged from 13 to 33 mm.
The LVOT becomes obstructed in late systole due to asymmetric hypertrophy of the septum, which generates a Venturi effect, leading to the dragging of the anterior mitral into the LVOT, a condition known as systolic anterior motion (SAM) of the mitral valve (Figure 2). Mitral regurgitation can occur when the AML is dragged towards the LVOT, by causing a space to appear between the mitral valve’s two leaflets. The mitral valve leaflets are elongated in HCM, especially the AML, which can be >30 mm long and thus be implicated in LVOT obstruction due to a steeper angle established between the AML and the septum. In 10% of HCM cases, there is a mid-cavity obstruction, and 25% of these patients develop LV apical aneurysm.
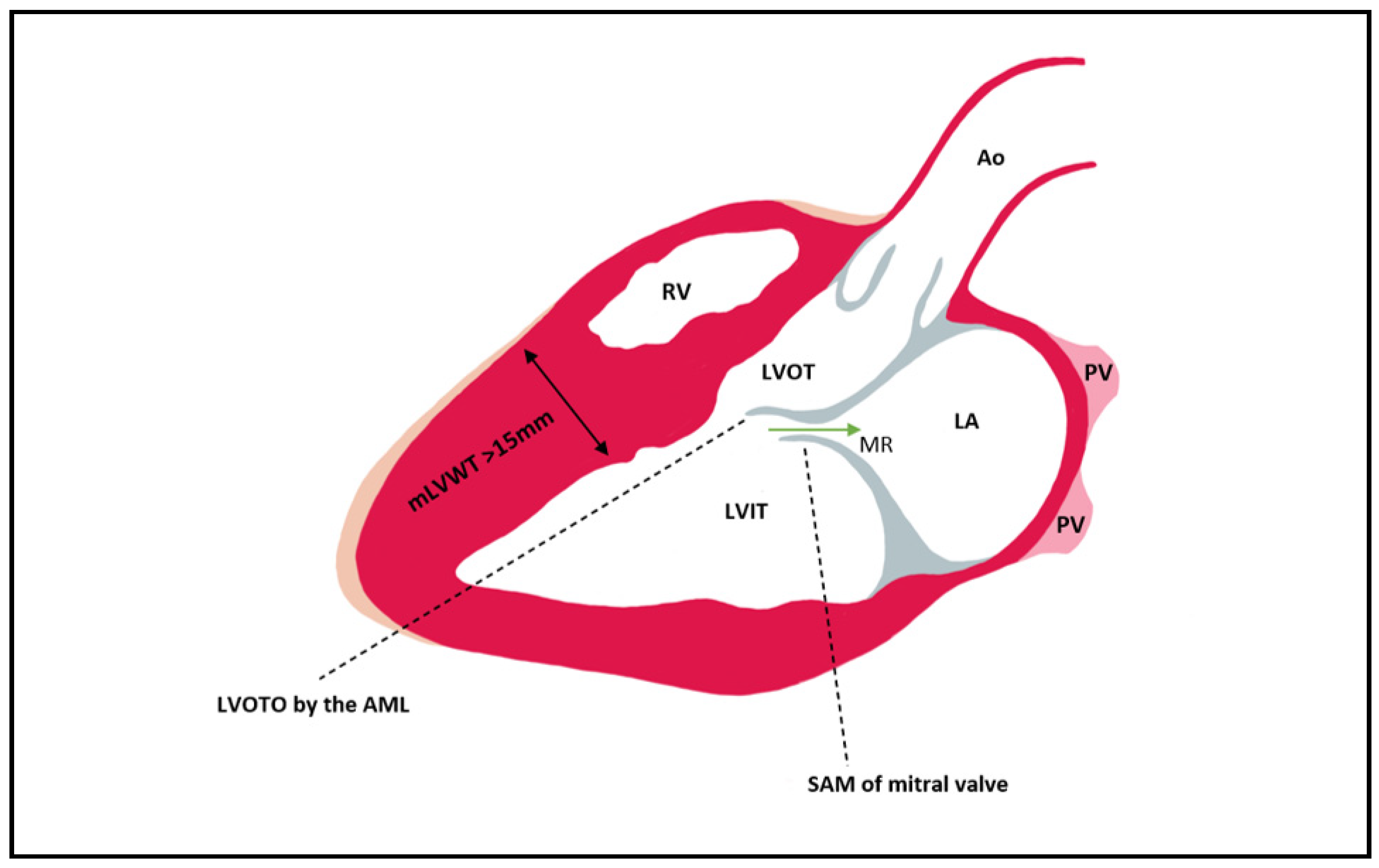
Figure 2. Left ventricular outflow tract obstruction by the anterior mitral leaflet which is pulled during late systole into the LVOT as a consequence of the Venturi effect. In most cases of HCM, the hypertrophy is mainly seen at the level of the septum. Mitral regurgitation can be present due to the SAM of AML that leads to a gap between the AML and PML (its presence can be established via Doppler). Ao = aorta; PV = pulmonary vein; LA = left atrium; LVIT = left ventricle inflow tract; LVOT = left ventricle outflow tract; RV = right ventricle; MR = mitral regurgitation; mLVWT = maximal Left ventricular wall thickness; LVOTO = Left ventricular outflow tract obstruction; SAM = Systolic anterior motion; AML = anterior mitral leaflet; PML = posterior mitral leaflet.
In a study on 1532 patients conducted by McLeod et al. [15] to better understand the cardiac features of this congenital disease, histological features identified included hyperchromatic nuclei in larger myocytes, myofibrillar disarray, interstitial and endocardial fibrosis. The curvature of the septum was used to classify the types of HCM: sigmoid septal morphology, reverse curve septal morphology, apical variant HCM, and neutral septal shape, with a weak association between the type of HCM and the degree of histological abnormality. Endocardial hypertrophy or mural plaque formation in the left ventricular outflow tract, as well as thickness of the anterior mitral leaflet, were characterized as hallmarks of HCM hearts that contributed to sudden death by Kocovski et al. [16]. According to Etfthimiadis et al. [17] outflow tract obstruction can occur not only in the subaortic area, but also in the mid-ventricular region (8%), resulting in an hourglass-shaped ventricle in 42.2% of patients. Overall, 26.5% of patients with midventricular obstruction had an apical aneurysm as a result of the elevated intracavitary pressure, and all of the obstructive HCM hearts’ left atria were dilated.
Hypertrophy was always present in Lamke’s study [18] of 204 septal myectomy HCM patients, with 100 of the 104 hearts exhibiting a significant subaortic septal bulge and four having an angled ventricular septum. In 79% of patients, myocardial disarray was present, and was more common in those under 65 years old. Fibrosis was found in 93% of the examined hearts, with 46% having abnormally thickened arteries. Galati et al. [19] observed three types of fibrosis: replacement fibrosis (53.3%), perimyocyte fibrosis (13.3%), and mixed (33.3%). The most common pattern was localized on the midwall, sometimes seen extending to the subepicardium or subendocardium. The upper anterior septum (36%) was the thickest region detected on echocardiography in Shapiro’s study [20], followed by the lower anterior septum (20%), upper posterior septum (9%), and the upper or lower free wall (21%).
3. Cardiac MR for HCM
Although echocardiography is an important tool in the evaluation of patients with left ventricular hypertrophy, it has certain limitations, especially in patients with an inadequate echocardiographic window. CMR bears an advantage in its capability to produce a more detailed three-dimensional image of the heart with high spatial and temporal resolution of the left ventricle. Additionally, it can differentiate between forms of HCM (Figure 3). The three main phenotypes of HCM are: asymmetric, concentric, and apical hypertrophy, with asymmetric septal being the most frequent between them. CMR can distinguish between different types of HCM. Moreover, CMR permits the visualization of the LV apex even in individuals with severe obesity.
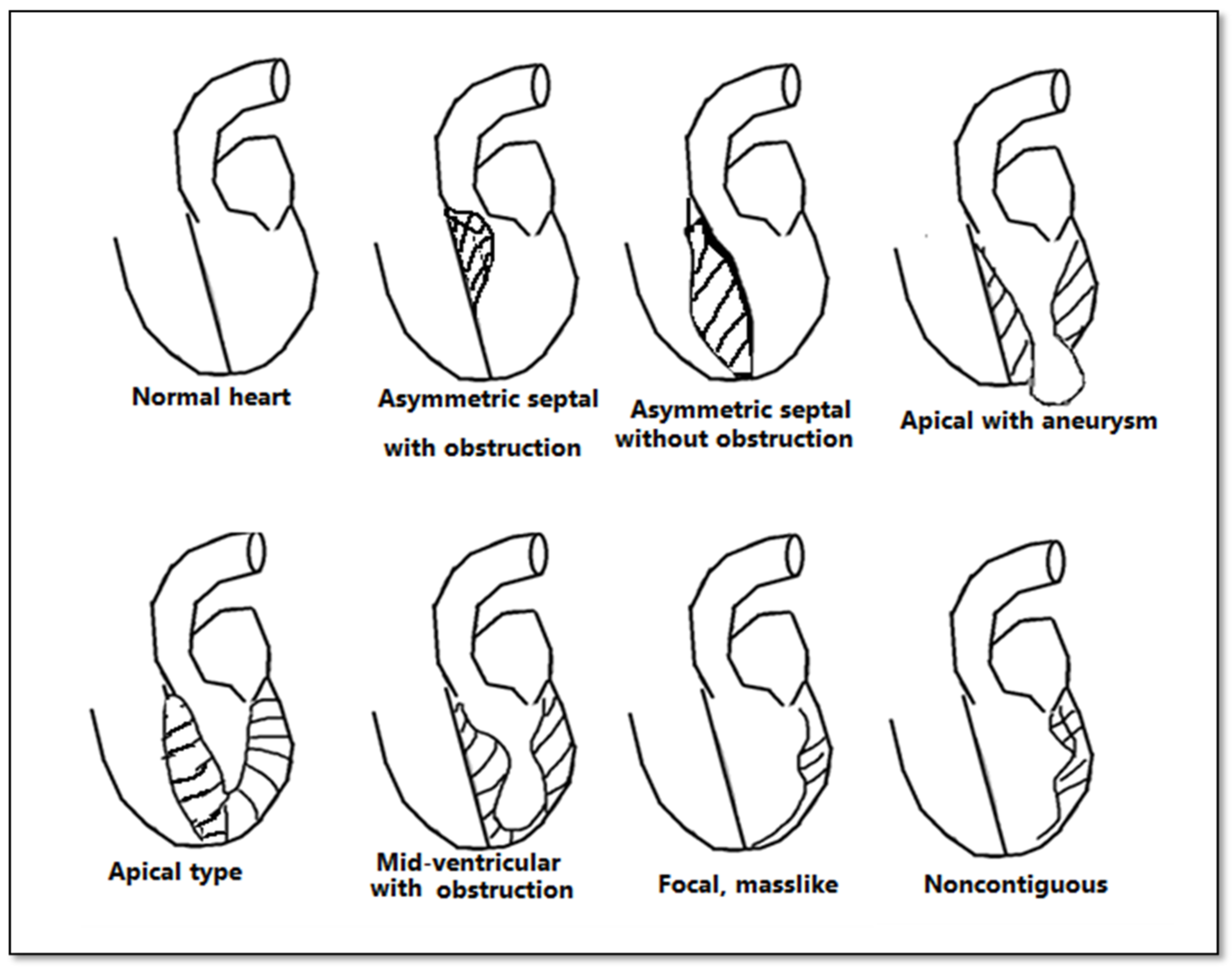
Figure 3. Anatomical types of HCM in function of the distribution of hypertrophy.
CMR remains the gold standard for measurement of left ventricular volumes as well as systolic and diastolic functions. Furthermore, CMR presents the advantage of being able to reliably characterize myocardial tissue. A maximum LV wall thickness higher than or equal to 15 mm in the end-diastolic phase is the most common diagnostic criteria for HCM. The relationship between late gadolinium enhancement and fibrotic zones has been studied extensively and has been associated with poorer clinical outcomes. Recently, T1 mapping techniques have improved, allowing for more detailed characterization of the diffuse fibrosis. For late gadolinium enhancement, 0.2 mmol/kg gadolinium contrast is injected at a speed of 1.5 mL/s. Ten minutes after infusion, acquisition begins, scanning the whole left ventricle from base to apex. Some categories of patients with HCM have a contraindication for CMR (Table 1). Myocardial fibrosis in HCM can be quantified as a percentage of the LV myocardium by using contrast-enhanced cardiac magnetic resonance with late gadolinium enhancement. Individuals with LGE >15% are at greater risk for sudden death [21][22][23].
Table 1. Contraindications for CMR.
|
Implanted ICDs * |
|
implanted pacemakers * |
|
brain ferromagnetic clips |
|
cochlear implants |
|
metal foreign body (bullet fragments, metallic splinter in the eye) |
|
claustrophobia |
* CMR has a relative contraindication in patients with an implanted cardiac device. MR Safe or MR Conditional devices, on the other hand, can be scanned using the proper protocol.
Until recently, there were no methods to determine myocardial disarray in vivo, but this can now be assessed using diffusion tensor cardiac magnetic resonance (DT-CMR) imaging. The preferential diffusion of water along cardiac muscle fibers in HCM can be detected and compared to control cases, with the diffusion being quantified as fractional anisotropy [11]. HCM shows lower diastolic fractional anisotropy than control patients according to Ariga R et al. [24].
4. CMR Differential Diagnosis of Thickened Myocardium
4.1. Differential Diagnosis with Athlete’s Heart
Athlete’s heart refers to morphologic modifications of the heart caused by intense physical activity: increased LV mass, increased LV diastolic diameter, and LV wall thickness. The absence of areas of delayed enhancement of the LV myocardium is a key aspect of the cardiac remodeling seen in athletes.
Athletes may acquire ventricular hypertrophy because of regular training, and it can be difficult to distinguish from HCM [25]. Histologically, the athlete’s cardiac hypertrophy is the result of cellular hypertrophy, whereas HCM is characterized by cellular disarray and extracellular expansion. When ECG and echocardiogram are inconclusive, CMR is an effective technique for distinguishing HCM from athlete’s heart, especially in patients with poor acoustic windows and hypertrophy affecting the apex, the basal antero-lateral wall, the inferior septum, or the inferior RV.
Luijkx et al. [26] conducted a study with the purpose of defining the characteristics of athlete’s heart and HCM, even when borderline hypertrophy (septal wall thickness values between 12 and 16 mm in males and 11 and 16 mm in women) was present. The researchers performed a retrospective study in which they examined various types of athletes and patients with HCM, considering LV diameters and ECV. The authors established a formula that can distinguish 99.5 percent of HCM patients from athletes’ heart (AUC = 0.995). As a result, as compared to both athletic and non-athletic healthy controls, the ratio of LV EDV (End Diastolic Volume) to LV EDM (End Diastolic Mass) was lower in patients with HCM. This model was still effective for borderline individuals (AUC = 0.992). This algorithm can be used to predict the progression of ventricular hypertrophy in various groups. Disproportional hypertrophy is a feature of HCM, with EDM growing as EDV decreases. Physiological hypertrophy, such as that seen in athletes, is proportionate, with both the EDM and EDV growing at the same time, resulting in increased heart strength and the ability to pump efficiently even under physical exertion.
4.2. Differential Diagnosis with Hypertensive Heart Disease
Hypertensive heart disease (HHD) is a disease that develops because of high blood pressure persisting for a long period of time. In contrast to HCM, which is characterized by myocyte disarray and focal interstitial fibrosis, hypertensive heart disease is defined by left ventricular hypertrophy, proportionate hyperplasia, and diffuse interstitial fibrosis. Since both diseases have concentric asymmetric left ventricle hypertrophy (LVWT 15 mm for HCM and 12 mm for HHD), LGE, reduced LV strain, and diastolic dysfunction, the type and quantity of fibrosis is the only way to differentiate them.
The effectiveness of T1 mapping for discriminating between HHD and HCM was investigated in a double-blinded retrospective study by Neisus et al. [27]. They examined 232 individuals, 108 of whom had been diagnosed with HCM and 53 with HHD. Patients with HCM and HHD were matched by gender, presence of LVH, and maximal LVWT, whereas those with similar age, LV mass index, and global and septal T1 were grouped together. In HCM patients, global native T1 mapping was significantly higher than global native T1 (108,631 vs. 110,439 ms). Global native T1 and LV mass index or maximal LVWT were mildly correlated only in HCM patients.
Takeda et al. [28] conducted a retrospective study to investigate the use of CMR to differentiate between forms of cardiomyopathies. Twenty-six participants underwent CMR, including Cine-MRI and LGE, as part of the study. Six were identified with cardiac amyloidosis, nine with end-stage HCM, and eleven with HHD. Patients with HHD had a higher LVEF than those with HCM, and pericardial effusions were more frequently seen in amyloidosis. In end-stage HCM, the number of LGE segments was higher than in HHD. LGE patterns in HCM were mostly observed in the anteroseptal or inferoseptal segments and they were mostly found to have patchy midwall or patchy epicardial patterns. On the other hand, midwall linear, midwall or epicardial LGE patterns were the most common in HHD, and were generally located in the septal to inferior areas of the midventricular level. The most useful type of CMR for establishing the diagnosis was Cine-MRI. LVEF was increased in HCM, but HHD was defined by progressive systolic dysfunction and a dilated LV cavity.
4.3. Differential Diagnosis with Infiltrative Cardiomyopathies
Restrictive cardiomyopathies such as amyloidosis, Fabry disease, glycogen storage disorders, sarcoidosis, iron overload, and myocardial fibrosis are difficult to distinguish from HCM since they have similar clinical characteristics and are defined by LVH [29].
Amyloid infiltration of the ventricular wall causes hypertrophy. Amyloidosis can be differentiated from other restrictive cardiomyopathies such as HCM, HHD, and diastolic heart failure with LV hypertrophy using CMR. In contrast to amyloidosis, which has a normal or decreased ventricular contraction, HCM has a hyperdynamic contraction. In addition, amyloid fibrils tend to accumulate in the subendocardium, leading to interstitial expansion and the distinctive “zebra pattern” of LGE. Nam et al. [30]. conducted a retrospective blind study to assess CMR’s role in differentiating amyloidosis from HCM. A total of 46 amyloidosis patients, 30 HCM patients, and 10 asymptomatic subjects were included in the study. They were all exposed to CMR with and without gadolinium contrast injection. The native T1 blood pool of the myocardium in amyloidosis was the highest of the two groups, while the native T1 blood pool of the LV cavity was the lowest. In amyloidosis, ECV was also higher.
Although cardiac amyloidosis exhibits a distinct LGE pattern, this approach is often unavailable due to the presence of renal impairment in most amyloidosis patients. Therefore, Jung et al. examined various forms of tissue tracking analyses in order to determine the best native CMR approach for differentiating between amyloidosis and HCM. The retrospective study comprised of 54 patients diagnosed with amyloidosis, 40 HCM patients, and 30 healthy controls. The authors suggested specific CMR techniques for amyloidosis detection, with amyloidosis patients displaying relative apical sparing and much better ratios of wall thickness to LVEF. Moreover, T1 mapping was found to be a very effective first step in distinguishing these two diseases (AUC = 0.880).
In sarcoidosis CMR, provides a thorough assessment of cardiac structure and function, as well as an amplification of the mediastinal lymph nodes, allowing for a precise diagnosis. LGE is also evident, in cardiac sarcoidosis.
CMR may occasionally reveal another source of LV hypertrophy in HCM patients: glycogen storage disorders, the most prevalent of which is Fabry disease. This condition is characterized by a gradual intracellular accumulation of neutral glycosphingolipids, which can occasionally cause LVH, particularly when alpha-galactosidase A, the implied lysosomal enzyme, has only partial activity. Since the pattern of LGE in this condition, which represents sphingolipid accumulation and interstitial expansion due to myocardial fibrosis, differs from that in HCM or HHD, CMR can be employed to diagnose it.
5. CMR for Risk Stratification in Adults with HCM
Late gadolinium enhancement is currently not a risk classification criterion in European or American guidelines. In European Guidelines, HCM Risk-SCD Calculator accounts for factors such as age, presence of LV hypertrophy, left atrial size, LV outflow tract gradient, family history of SCD, nonsustained ventricular tachycardia, and idiopathic syncope [31]. In American guidelines, personal history of aborted SCD, ventricular fibrillation, or sustained ventricular tachycardia; family history of SCD; and syncope are all considered relevant risk factors [32]. Nonsustained ventricular tachycardia on monitoring, severe increase in left ventricular (LV) wall thickness (>30 mm), and hypotensive response to exercise are all markers with less convincing evidence.
However, in recent years, LGE has emerged as a risk marker for unfavorable HCM outcomes. LGE may be present in around half of all HCM patients. Nevertheless, the presence of LGE cannot be considered as a sole indicator of an ICD. The extent of LGE may be a more powerful indicator than its mere presence. A study [33] on 1293 patients with HCM followed for 3.3 years found that not just the presence of LGE but the extent of >15% of LV mass was associated with a two-fold increase in the probability of SCD.
6. Special Considerations in Children
HCM is responsible for 42% of cardiomyopathy in children, and annual HCM-related mortality is close to 1% [34][35]. In children and young individuals, HCM is a common cause of arrhythmias and sudden cardiac death [36]. Pediatric HCM encompasses a more diverse group of abnormalities when compared with adult HCM. HCMs with sarcomeric gene abnormalities are called primary HCMs, and non-sarcomeric gene abnormalities are called secondary HCMs.
Most of them, 40–60%, are due to mutations in the sarcomeric genes. Other causes of HCM include inherited metabolic errors such as glycogen storage disorders (Pompe disease, Danon disease, Cori-Forbes disease, PRKAG2 syndrome), lysosomal storage diseases (mucopolysaccharidoses), fatty acid oxidation disorders, endocrine disorders, neuromuscular diseases, mitochondrial diseases (Barth syndrome, Friedreich ataxia), and malformation syndromes (Noonan syndrome, Costello syndrome, cardiofaciocutaneous syndrome, neurofibromatosis Type 1, Legius Syndrome) which account for up to 35% HCM cases in children [37][38].
While echocardiography remains the gold standard for diagnosing HCM in children, cardiovascular magnetic resonance imaging can be useful in pediatric patients with diagnostic uncertainty, such as those with a suspected metabolic or lysosomal storage disorder or malformation syndrome, in case of poor echocardiographic imaging windows, or as an adjunct in assessing the risk of sudden cardiac death by assessing late gadolinium enhancement in children with confirmed HCM [39].
References
- Prinz, C.; Farr, M.; Hering, D.; Horstkotte, D.; Faber, L. The Diagnosis and Treatment of Hypertrophic Cardiomyopathy. Dtsch. Ärzteblatt Int. 2011, 108, 209–215.
- Motevali, M.; Siahi, Z.; Mohammadzadeh, A.; Sangi, A. Cardiac Magnetic Resonance Imaging (MRI) Findings in Arrhythmogenic Right Ventricular Dysplasia (ARVD) Compared with Echocardiography. Med. Sci. 2018, 6, 80.
- Lopes, L.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype–phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811.
- Akhtar, M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pr. 2018, 2018, 36.
- Richard, P.; Charron, P.; Carrier, L. Hypertrophic cardiomyopathy. Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. ACC Curr. J. Rev. 2003, 12, 60.
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto Hypertrophic Cardiomyopathy Genotype Score for Prediction of a Positive Genotype in Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26.
- Hindieh, W.; Chan, R.; Rakowski, H. Complementary Role of Echocardiography and Cardiac Magnetic Resonance in Hypertrophic Cardiomyopathy. Curr. Cardiol. Rep. 2017, 19, 81.
- Reichek, N. Imaging cardiac morphology in hypertrophic cardiomyopathy: Recent advances. Curr. Opin. Cardiol. 2015, 30, 461–467.
- Varma, P.; Neema, P.; Pk, V. Hypertrophic cardiomyopathy: Part 1—Introduction, pathology and pathophysiology. Ann. Card. Anaesth. 2014, 17, 118.
- Makavos, G.; Κairis, C.; Tselegkidi, M.-E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic cardiomyopathy: An updated review on diagnosis, prognosis, and treatment. Heart Fail. Rev. 2019, 24, 439–459.
- Habib, M.; Hoss, S.; Rakowski, H. Evaluation of Hypertrophic Cardiomyopathy: Newer Echo and MRI Approaches. Curr. Cardiol. Rep. 2019, 21, 75.
- Teare, D. Asymmetrical hypertrophy of the heart in young adults. Br. Heart J. 1958, 20, 1–8.
- Sutton, M.G.S.J.; Lie, J.T.; Anderson, K.R.; O’Brien, P.C.; Frye, R.L. Histopathological specificity of hypertrophic obstructive cardiomyopathy. Myocardial fibre disarray and myocardial fibrosis. Heart 1980, 44, 433–443.
- Maron, B.J.; Mackey-Bojack, S.; Facile, E.; Duncanson, E.; Rowin, E.J.; Maron, M.S. Hypertrophic Cardiomyopathy and Sudden Death Initially Identified at Autopsy. Am. J. Cardiol. 2020, 127, 139–141.
- McLeod, C.J.; Ommen, S.R.; Ackerman, M.J.; Weivoda, P.L.; Shen, W.K.; Dearani, J.A.; Schaff, H.; Tajik, A.J.; Gersh, B.J. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2007, 28, 2583–2588.
- Kocovski, L.; Fernandes, J.; Cardiac, S. Death: A Modern Pathology Approach to Hypertrophic Cardiomyopathy. Arch. Pathol. Lab. Med. 2015, 139, 413–416.
- Efthimiadis, G.K.; Pagourelias, E.D.; Parcharidou, D.; Gossios, T.; Kamperidis, V.; Theofilogiannakos, E.K.; Pappa, Z.; Meditskou, S.; Hadjimiltiades, S.; Pliakos, C.; et al. Clinical Characteristics and Natural History of Hypertrophic Cardiomyopathy With Midventricular Obstruction. Circ. J. 2013, 77, 2366–2374.
- Lamke, G.T.; Allen, R.D.; Edwards, W.D.; Tazelaar, H.D.; Danielson, G.K. Surgical pathology of subaortic septal myectomy associated with hypertrophic cardiomyopathy. Cardiovasc. Pathol. 2003, 12, 149–158.
- Galati, G.; Leone, O.; Pasquale, F.; Olivotto, I.; Biagini, E.; Grigioni, F.; Pilato, E.; Lorenzini, M.; Corti, B.; Foà, A.; et al. Histological and Histometric Characterization of Myocardial Fibrosis in End-Stage Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2016, 9, e003090.
- Shapiro, L.M.; McKenna, W.J. Distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: A two-dimensional echocardiographic study. J. Am. Coll. Cardiol. 1983, 2, 437–444.
- Hensley, N.; Dietrich, J.; Nyhan, D.; Mitter, N.; Yee, M.S.; Brady, M. Hypertrophic cardiomyopathy: A review. Anesth. Analg. 2015, 120, 554–569.
- To, A.C.; Dhillon, A.; Desai, M.Y. Cardiac magnetic resonance in hypertrophic cardiomyopathy. JACC Cardiovasc. Imaging 2011, 4, 1123–1137.
- Rowin, E.J.; Maron, M.S. The Role of Cardiac MRI in the Diagnosis and Risk Stratification of Hypertrophic Cardiomyopathy. Arrhythmia Electrophysiol. Rev. 2016, 5, 197–202.
- Ariga, R.; Tunnicliffe, E.M.; Manohar, S.; Mahmod, M.; Raman, B.; Piechnik, S.K.; Francis, J.M.; Robson, M.D.; Neubauer, S.; Watkins, H. Identification of Myocardial Disarray in Patients With Hypertrophic Cardiomyopathy and Ventricular Arrhythmias. J. Am. Coll. Cardiol. 2019, 73, 2493–2502.
- Sharma, S.; Elliott, P.; Whyte, G.; Mahon, N.; Virdee, M.S.; Mist, B.; McKenna, W.J. Utility of metabolic exercise testing in distinguishing hypertrophic cardiomyopathy from physiologic left ventricular hypertrophy in athletes. J. Am. Coll. Cardiol. 2000, 36, 864–870.
- Luijkx, T.; Cramer, M.J.; Buckens, C.F.; Zaidi, A.; Rienks, R.; Mosterd, A.; Prakken, N.; Dijkman, B.; Mali, W.P.T.M.; Velthuis, B.K. Unravelling the grey zone: Cardiac MRI volume to wall mass ratio to differentiate hypertrophic cardiomyopathy and the athlete’s heart. Br. J. Sports Med. 2013, 49, 1404–1409.
- Neisius, U.; El-Rewaidy, H.; Nakamori, S.; Rodriguez, J.; Manning, W.J.; Nezafat, R. Radiomic Analysis of Myocardial Native T1 Imaging Discriminates Between Hypertensive Heart Disease and Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2019, 12, 1946–1954.
- Takeda, M.; Amano, Y.; Tachi, M.; Tani, H.; Mizuno, K.; Kumita, S. MRI differentiation of cardiomyopathy showing left ventricular hypertrophy and heart failure: Differentiation between cardiac amyloidosis, hypertrophic cardiomyopathy, and hypertensive heart disease. Jpn. J. Radiol. 2013, 31, 693–700.
- O’Hanlon, R.; Pennell, D.J. Cardiovascular Magnetic Resonance in the Evaluation of Hypertrophic and Infiltrative Cardiomyopathies. Heart Fail. Clin. 2009, 5, 369–387.
- Nam, B.-D.; Kim, S.M.; Na Jung, H.; Kim, Y.; Choe, Y.H. Comparison of quantitative imaging parameters using cardiovascular magnetic resonance between cardiac amyloidosis and hypertrophic cardiomyopathy: Inversion time scout versus T1 mapping. Int. J. Cardiovasc. Imaging 2018, 34, 1769–1777.
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2013, 35, 2010–2020.
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2011, 58, 2703–2738.
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Pencina, M.J.; Assenza, G.E.; Haas, T.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation 2014, 130, 484–495.
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; Lowe, A.M.; Orav, E.J.; Cox, G.F.; Lurie, P.R.; McCoy, K.L.; McDonald, M.A.; Messere, J.E.; et al. The Incidence of Pediatric Cardiomyopathy in Two Regions of the United States. N. Engl. J. Med. 2003, 348, 1647–1655.
- Colan, S.D.; Lipshultz, S.E.; Lowe, A.M.; Sleeper, L.A.; Messere, J.; Cox, G.F.; Lurie, P.R.; Orav, E.J.; Towbin, J.A. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007, 115, 773–781.
- Chaowu, Y.; Shihua, Z.; Jian, L.; Li, L.; Wei, F. Cardiovascular Magnetic Resonance Characteristics in Children with Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2013, 6, 1013–1020.
- Kindel, S.J.; Miller, E.M.; Gupta, R.; Cripe, L.H.; Hinton, R.B.; Spicer, R.L.; Towbin, J.A.; Ware, S.M. Pediatric Cardiomyopathy: Importance of Genetic and Metabolic Evaluation. J. Card. Fail. 2012, 18, 396–403.
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic value of LGE-CMR in HCM: A meta-analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402.
- Decker, J.A.; Rossano, J.W.; Smith, E.O.; Cannon, B.; Clunie, S.K.; Gates, C.; Jefferies, J.L.; Kim, J.J.; Price, J.F.; Dreyer, W.J.; et al. Risk Factors and Mode of Death in Isolated Hypertrophic Cardiomyopathy in Children. J. Am. Coll. Cardiol. 2009, 54, 250–254.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.6K
Revisions:
2 times
(View History)
Update Date:
11 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No