Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Janusz Blasiak | + 2485 word(s) | 2485 | 2022-01-29 08:48:26 | | | |
| 2 | Catherine Yang | Meta information modification | 2485 | 2022-03-03 07:44:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Blasiak, J. Waste Clearing in Retinal Pigment Epithelium Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/20130 (accessed on 27 July 2026).
Blasiak J. Waste Clearing in Retinal Pigment Epithelium Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/20130. Accessed July 27, 2026.
Blasiak, Janusz. "Waste Clearing in Retinal Pigment Epithelium Cells" Encyclopedia, https://encyclopedia.pub/entry/20130 (accessed July 27, 2026).
Blasiak, J. (2022, March 03). Waste Clearing in Retinal Pigment Epithelium Cells. In Encyclopedia. https://encyclopedia.pub/entry/20130
Blasiak, Janusz. "Waste Clearing in Retinal Pigment Epithelium Cells." Encyclopedia. Web. 03 March, 2022.
Copy Citation
Waste clearing in retinal pigment epithelium (RPE) cells includes proteasomal degradation, heterophagy, macroautophagy and mitophagy. Exosomes can also be involved in waste removal in RPE cells. The ubiquitin proteasome system (UPS) is mainly responsible for degradation of damaged or no longer needed proteins. Autophagy can degrade damaged organelle and may also take a part in degradation proteins when other clearance processes are failed. RPE cells phagocytose used photoreceptors outer segments (POS) with their subsequent autophagy-lysosomal degradation.
age-related macular degeneration
autophagy
heterophagy
retinal pigment epithelium
proteasome
photoreceptor outer segments
unfolded protein response
1. Unfolded Protein Response and Endoplasmic Reticulum-Associated Degradation
Unfolded protein response is a signaling cascade activated in response to endoplasmic reticulum (ER) stress manifested by the accumulation of unfolded and damaged proteins. UPR may also be activated by damaged mitochondria. Expression of UPR genes increases in oxidative stress and it was shown that the stress caused by cigarette smoke extracts induced UPR in RPE cells [1][2]. In ER UPR is activated by pathways initiated by three different sensors: protein kinase-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (Figure 1) [3][4][5][6][7]. Oxidative stress enhances the transcription of these proteins in RPE cells [2]. The UPR signalling activates transcription factors and protein kinases leading to an adaptive response involving the activation of proteasomal degradation and autophagy, chaperone induction and enhancement of antioxidant defence [8][9]. Inefficient attempts to restore homeostasis cause UPR to induce cell death by apoptosis. UPR increases apoptosis in RPE cells treated with cigarette smoke extract [10][11][12]. Therefore, UPR can play a role in AMD pathogenesis as it is involved in detecting of improper proteins and their degradation in RPE.
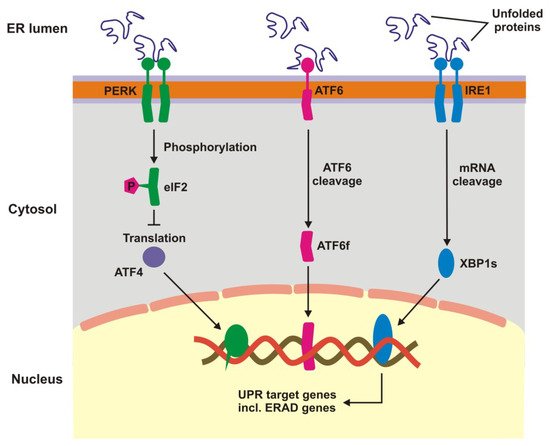
Figure 1. Unfolded protein response. When unfolded, misfolded and damaged proteins accumulate in endoplasmic reticulum (ER), they can induce unfolded protein response (UPR), a signaling cascade with the involvement of protein kinase-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). This cascade leads to a stop in translation of faulty proteins, degradation of misfolded proteins and increased synthesis of chaperons involved in protein folding. If these mechanisms fall, UPR switch to pro-apoptotic response. XBP1s—X-box binding protein 1 specificity protein, eIF2—translation initiation factor 2, ERAD—ER-associated degradation, ATF6f—the transcriptional activator domain of ATF6, P—phosphate residue.
2. Ubiquitin Proteasome System
The process of UPS degradation is initiated by specific enzymes, E1–E3, that attach ubiquitin to the substrate in an ATP-dependent manner. Substrates with polyubiquitin tail are transported to proteasome where they undergo degradation.
Typical 26S proteosomal complex contains three domains: a core particle (20S) with two regulatory particles (19S, caps, lids) [15]. Regulatory particles are involved in the recognition and binding of polyubiquitinated proteins and their transport to the catalytic core located at the inner surface of the 20S subunit. The core particle is composed of four heptameric annular complexes—two outer α subunits, which play a structural function and two inner β subunits with catalytic activity. The β subunits contain proteolytic active sites located on proteasomal interior surface. Energy from ATP hydrolysis is used to open lid, unfold polyubiquitinated proteins and their transport inside 20S subunit.
Oxidative stress, a major factor of AMD pathogenesis, is associated with an increased production of cellular waste, but on the other hand it may damage components of cellular waste clearing systems, which can contribute to AMD progression. A mild oxidative stress can regulate UPS activity through the stimulation of E1 and E2 to bind ubiquitin as well as the 26S proteasome [16][17]. However, strong oxidative stress can damage E1 and E2, blocking ubiquitin binding. This effect can be associated with a high concentration of oxidized glutathione, competing with E1 and E2 on their binding sites in ubiquitin [18]. Oxidative stress could also directly inactivate 26S proteasome through detachment of the 19S subunit from 20S core particle [10][19].
Reduction of proteasome activity during lifetime is associated with aging, another critical factor in AMD pathogenesis. Aging results in reduced expression of UPS genes and may lead to the collapse of proteasome complex, resulting in an accumulation of cellular debris [20][21][22][23][24]. These effects are correlated with weakening of the sustaining activity of the proteins quality control system and especially chaperone proteins, which play a significant role in UPS [25][26][27]. Several other effects can underline lowering of the efficacy of UPS with age, including alternations in the composition of the proteasomal subunits, reduced stability of proteasomes or their inactivation [28][23][24][25]. It has been shown that during replicative senescence the level of the β subunits decreases [29]. Proteasome activity is associated with the rate of cellular aging and the entry of cells into the senescence pathway [30]. Moreover, decreasing activity of proteasome results in increase of damaged proteins of the respiratory chain, resulting in mitochondrial dysfunction and an increase of cellular ROS level [31][32].
Insufficient activity of UPS leads to the escalation of protein deposits followed by an increase in the level of ROS and induction of chronic inflammation. Inflammation combined with constantly weakening waste cleaning system in RPE cells can induce their senescence, resulting in the development and progression of AMD [33].
Liu et al. showed that photooxidative stress decreased the activity of UPS in RPE. This interaction increased the expression of genes encoding proinflammatory interleukins 6 and 8 (IL-6 and -8) and downregulated the anti-inflammatory genes MCP-1 (monocyte chemoattractant protein-1) and CFH (complement factor H) [34]. Similar results obtained Qin et al. who showed that a decrease in proteasome activity in RPE led to dysregulation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signalling pathway [35]. It was demonstrated in a mouse model that dysregulation of UPS led to retinal degeneration through photoreceptor cell death by apoptosis in a caspase-independent pathway [36]. De Carvalho. et al. suggested that proteasomal regulation may play a significant role in the control of neovascularization process important in wet AMD [37]. They showed an efficient action of UPS counteracting degenerative changes in ARPE-19 (human retinal pigment epithelial) cells through the control of the TGFβ (transforming growth factor-β) signalling.
3. Autophagy
Autophagy degrades damaged or unneeded proteins in lysosomes. Many proteins are involved in this process, including autophagy related proteins (ATGs), mechanistic target of rapamycin (mTOR), the serine/threonine uncoordinated-51-like kinases 1 and 2 (ULK1 and ULK2), FIP-200, p62/SQSTM1, microtubule-associated protein light chain 3 (LC3) and others. Several modes of autophagy can function, including macroautophagy (usually referred as to autophagy), chaperone-mediated autophagy and microautophagy.
Autophagy is initiated by the formation of a double-membrane vesicle, autophagosome, enclosing material to be degraded (cargo) that is delivered to the lysosome, where degradation and recycling occur [38].
Autophagy impairment, caused by the depletion of the core autophagy genes ATG5 and ATG7, was associated with an AMD-like phenotype in mouse RPE cells. This phenotype was manifested by RPE thickening, hypertrophy or hypotrophy, pigmentary abnormalities and accumulation of oxidized proteins [39]. A2E, the main hydrophobic constituent of lipofuscin can induce damage to RPE cells through the inhibition of autophagy [40]. These findings suggest that autophagy prevents detrimental effects of A2E and inhibits the production of inflammatory factors in RPE.
Many reports imply that oxidative stress induces autophagy in RPE cells [41][42][43][44]. Studies on RPE cells from AMD donors and mice with AMD-like phenotype suggest that autophagy increases during aging and AMD [42]. However, the autophagosomes formation in late AMD was reported to occur at lower rate than in early stages. This study also revealed that chronic oxidative stress decreased autophagic flux. Autophagy can prevent retinal cells from the damaging effects of oxidative stress [41][42]. Rapamycin induced autophagy in RPE cells and led to reduced accumulation of lipofuscin, whereas leupeptin, a blocker of autophagy, caused an increase in lipofuscin formation [42].
Autophagy can protect RPE cells from cell death induced by oxidative stress. RPE cells treated with paraquat, an inducer of oxidative stress and cultured with autophagy inhibitor 3-methyladenine (3-MA) showed an increase in the number of apoptotic cells compared to cells with undisturbed autophagy [41]. RPE cells under oxidative stress increased the expression of p62/SQSTM1 and autophagy [43]. Rotenone, an agent inducing mitotic catastrophe, increased autophagy and mitophagy in RPE cells protecting these cells from death [44].
The FIP200 protein is important in autophagy induction as it is involved in the formation of autophagosomes [45]. The conditional knockout of gene encoding FIP200 (FIP200 cKO) in mice resulted in a reduction of autophagy. These animals also displayed changes in the phenotype of RPE cells, including lipid accumulation, increasing with age. The reduction of autophagy in FIP200 cKO mice led to photoreceptors loss and retinal dysfunction [46].
Mice with knockout of ATP-binding cassette subfamily A member 4 (Abca4) and retinol dehydrogenase 8 (Rdh8) genes are characterized by impaired clearing of atRAL and they were a model of light-induced retinal degeneration [47]. Retinas of these mice were characterized by a delay in atRAL removal after light exposure [48]. Additionally, light illumination led to an increased expression of the LC3B-II and PARKIN proteins, a marker of autophagosome formation and a mitophagy regulator, respectively. These results suggest that autophagy plays an important role in protecting the retina from damage caused by light.
Injection of amyloid-β, which is a main component of drusen, to murine vitreous resulted in a upregulation of autophagy markers LC3, ATG5 and BECLIN-1. Human RPE cells treated with amyloid-β also showed autophagy induction and upregulated expression of cytokines [49].
4. Mitophagy
Damaged mitochondria are removed in a highly specific and selective pathway called mitophagy. All mechanistic aspects of this process are not exactly known and several models of it have been presented [50].
In a model proposed by Ding and Yin mitophagy is a two-steps process involving induction of canonical autophagy with ATG proteins and priming of mitochondria [51]. Canonical autophagy is underlined by several mechanisms, including AMPK activation induced by ATP depletion and suppression of mTOR mediated by mitochondrial damage resulting in ROS overproduction. These ROS induce further mitochondrial damage, which amplifies the inducing signal. Mitochondria priming could be PARKIN-dependent or independent. In the former, depolarization of mitochondrial membrane results in compromised cleavage of the PINK1 (PTEN (phosphatase and tensin homolog) induced kinase 1) protein mediated by the mitochondrial rhomboid protease PARL (presenilins-associated rhomboid-like protein, mitochondrial). Stabile PINK1 recruits PARKIN to mitochondria resulting in subsequent ubiquitination of proteins localized on the outer mitochondrial membrane. These proteins can be degraded by UPS or bound by p62/SQSTM1, which directly interacts with LC3 to bind autophagosome to faulty mitochondria. Selective mitophagy can be supported by the PI3K (phosphoinositide 3-kinase) complex activated by Ambra1. An enhanced expression of the FUNDC1 (FUN14 domain containing 1) and BNIP3L (BCL2 (B-cell lymphoma 2) interacting protein 3 like) proteins in impaired mitochondria may occur in the PARKIN-independent pathway of mitophagy. These proteins induce autophagosome to target mitochondria by a direct interaction with LC3. In this pathway, damaged mitochondria can be also targeted by Smurf1 (SMAD specific E3 ubiquitin protein ligase 1) to ubiquitinate mitochondrial proteins and induce mitophagy. ULK1 can phosphorylate ATG13 upon activation by Hsp90 (heat shock protein 90 kDa) to promote mitophagy. PINK1 is cleaved by mitochondrial proteases and degraded in the proteasome [52]. PINK1 activates PARKIN by its phosphorylation at S65 [53]. PINK1 targets the same residue in phosphorylation of ubiquitin in the S65 position [54]. Several other mechanisms of mitophagy, both PARKIN-dependent and independent could be considered.
Aging reduces the efficacy of mitophagy, which leads to the accumulation of damaged mitochondria (reviewed in [52]). Aged RPE cells have more mitochondrial DNA (mtDNA) damage and display a decreased ability to repair it as compared to young RPE [55][56][57][58]. The number of mitochondria decreased with age in rhesus RPE and aging mitochondria had an increased length and formed clusters [55].
5. Exosomal Degradation
Exosomes are extracellular vehicles released by various cells, including epithelial cells [59]. They are an important element of the cell-to-cell communication (reviewed in [60]). They can carry out of the cells various molecules, including peptides, proteins, lipids, RNA and DNA, so they can be also considered as an important element of cellular waste clearing. In fact, waste elimination function was attributed to exosomes earlier than their communicative potential. However, some of molecules exported from cells by exosome can be only carriers of biological information. Exosome is a 9–11 protein complex having, similarly to proteasome, ring-like core structure that in humans contains nine subunits [61]. Core proteins of eukaryotic exosomes display RNase activity and belong to the RNase PH class [62]. Exosomes are present and display activity in the cytoplasm, nucleus and nucleolus. RNA degradation by exosomes is their best known and likely the main function. To perform it, the core exosomes display both exo- and endoribonuclease activities. Many aspects of exosome functioning, both as a cellular garbage bin and as an important element of the cell-to-cell communication, need further research. It is worth noting that exosomes can cooperate with autophagy in cellular waste clearing [63].
Some proteins which can be found in drusen, including annexin, enolase, CD63 are features of exosomes [64][65][66].
Blue light is an environmental AMD risk factor and it induces detrimental changes in the retina, which are associated with oxidative stress and overproduction of cellular waste. An increase in the proinflammatory molecules in the content of exosomes released by RPE cells after photoactive blue-light stimulation was observed [67]. Moreover, a higher level of the NLRP3 (NACHT (neuronal apoptosis inhibitor protein, class 2 transcription activator of the MHC, heterokaryon incompatibility and telomerase-associated protein 1), NLR (nucleotide-binding domain, leucine-rich repeat-containing family), and PYD (pyrin domain)-containing protein 3) inflammasome was observed in that study.
Emerging evidence suggests the role of exosomes in the activation of the complement in the immediate vicinity of RPE cells [68]. Exosomes were suggested to play a role in the occurrence and development of choroidal neovascularization, so they can be important in mechanisms of wet AMD pathogenesis and developing therapeutic strategies in this disease [69]. Exosomal proteins found in aqueous humor were postulated to be an independent molecular marker in wet AMD [70]. Exosomal miRNA, which can be important to stimulate target cells, was recently discovered in AMD [71]. Therefore, exosomes may play a multiple role in AMD pathogenesis, but uncovering the precise mechanism of this role requires further studies.
6. Heterophagy
Heterophagy, a digestion of extracellular material inside the cell, is intensively carried out in RPE cells as they constantly degrade POS to maintain the function of photoreceptors. Each RPE cell is challenged by digestion of POS from 30 to 40 photoreceptors [72].
Heterophagy in RPE cells involves the recognition and attachment of a POS discs, its digestion, the formation of phagosome and its fusion with lysosome and the final degradation [73]. Integrins, including ITGAV (integrin alpha V)-ITGB5 (integrin subunit beta 5) are necessary to bind POS, which ingestion requires the MERTK (c-mer proto-oncogene tyrosine kinase) protein [74]. After ingestion of extracellular cargo into vesicles they are transported to the basal end of the cell and fuse with lysosomes to degrade the cargo as it does in autophagy [75]. Heterophagy impairment can lead to accumulation of photoreceptor cell waste resulting in chronic inflammation. POS recognition by RPE may be a critical step in heterophagy as defects in this process results in death of photoreceptors [76].
References
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073.
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380.
- Bazi, A.; Keramati, M.R.; Gholamin, M. Role of Oxidative Stress in Modulating Unfolded Protein Response Activity in Chronic Myeloid Leukemia Cell Line. Iran. Biomed. J. 2016, 20, 63–67.
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radic. Biol. Med. 2014, 69, 1–14.
- Hong, M.; Li, M.; Mao, C.; Lee, A.S. Endoplasmic reticulum stress triggers an acute proteasome-dependent degradation of ATF6. J. Cell. Biochem. 2004, 92, 723–732.
- Hong, M.; Luo, S.; Baumeister, P.; Huang, J.M.; Gogia, R.K.; Li, M.; Lee, A.S. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J. Biol. Chem. 2004, 279, 11354–11363.
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348.
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231.
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438.
- Huang, C.; Wang, J.J.; Ma, J.H.; Jin, C.; Yu, Q.; Zhang, S.X. Activation of the UPR protects against cigarette smoke-induced RPE apoptosis through up-regulation of Nrf2. J. Biol. Chem. 2015, 290, 5367–5380.
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 2008, 45, 50–59.
- Tagawa, Y.; Hiramatsu, N.; Kato, H.; Sakoh, T.; Nakajima, S.; Hayakawa, K.; Saito, Y.; Johno, H.; Takahashi, S.; Gu, L.; et al. Induction of CCAAT/enhancer-binding protein-homologous protein by cigarette smoke through the superoxide anion-triggered PERK-eIF2alpha pathway. Toxicology 2011, 287, 105–112.
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR. Trends Biochem. Sci. 2018, 43, 593–605.
- McCaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235.
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36.
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R., Jr.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997, 272, 28218–28226.
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594.
- Obin, M.; Shang, F.; Gong, X.; Handelman, G.; Blumberg, J.; Taylor, A. Redox regulation of ubiquitin-conjugating enzymes: Mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998, 12, 561–569.
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997, 11, 526–534.
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 2010, 3, ra88.
- Bulteau, A.L.; Verbeke, P.; Petropoulos, I.; Chaffotte, A.F.; Friguet, B. Proteasome inhibition in glyoxal-treated fibroblasts and resistance of glycated glucose-6-phosphate dehydrogenase to 20 S proteasome degradation in vitro. J. Biol. Chem. 2001, 276, 45662–45668.
- Lee, C.W.; La Thangue, N.B. Promoter specificity and stability control of the p53-related protein p73. Oncogene 1999, 18, 4171–4181.
- Vernace, V.A.; Arnaud, L.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 2007, 21, 2672–2682.
- Vernace, V.A.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging and regulated protein degradation: Who has the UPPer hand? Aging Cell 2007, 6, 599–606.
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The stress of protein misfolding: From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530.
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 644–646.
- Crowe, E.; Sell, C.; Thomas, J.D.; Johannes, G.J.; Torres, C. Activation of proteasome by insulin-like growth factor-I may enhance clearance of oxidized proteins in the brain. Mech. Ageing Dev. 2009, 130, 793–800.
- Chondrogianni, N.; Gonos, E.S. Proteasome function determines cellular homeostasis and the rate of aging. Adv. Exp. Med. Biol. 2010, 694, 38–46.
- Ding, Q.; Dimayuga, E.; Keller, J.N. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid. Redox Signal. 2006, 8, 163–172.
- Ding, Q.; Dimayuga, E.; Markesbery, W.R.; Keller, J.N. Proteasome inhibition induces reversible impairments in protein synthesis. FASEB J. 2006, 20, 1055–1063.
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxid. Med. Cell. Longev. 2017, 2017, 5293258.
- Liu, Z.; Qin, T.; Zhou, J.; Taylor, A.; Sparrow, J.R.; Shang, F. Impairment of the ubiquitin-proteasome pathway in RPE alters the expression of inflammation related genes. Adv. Exp. Med. Biol. 2014, 801, 237–250.
- Qin, T.; Gao, S. Inhibition of Proteasome Activity Upregulates IL-6 Expression in RPE Cells through the Activation of P38 MAPKs. J. Ophthalmol. 2018, 2018, 5392432.
- Ando, R.; Noda, K.; Tomaru, U.; Kamoshita, M.; Ozawa, Y.; Notomi, S.; Hisatomi, T.; Noda, M.; Kanda, A.; Ishibashi, T.; et al. Decreased proteasomal activity causes photoreceptor degeneration in mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4682–4690.
- Ramos de Carvalho, J.E.; Verwoert, M.T.; Vogels, I.M.C.; Reits, E.A.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Involvement of the ubiquitin-proteasome system in the expression of extracellular matrix genes in retinal pigment epithelial cells. Biochem. Biophys. Rep. 2018, 13, 83–92.
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743.
- Zhang, Y.; Cross, S.D.; Stanton, J.B.; Marmorstein, A.D.; Le, Y.Z.; Marmorstein, L.Y. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol. Vis. 2017, 23, 228–241.
- Saadat, K.A.; Murakami, Y.; Tan, X.; Nomura, Y.; Yasukawa, T.; Okada, E.; Ikeda, Y.; Yanagi, Y. Inhibition of autophagy induces retinal pigment epithelial cell damage by the lipofuscin fluorophore A2E. FEBS Open Bio 2014, 4, 1007–1014.
- Baek, A.; Yoon, S.; Kim, J.; Baek, Y.M.; Park, H.; Lim, D.; Chung, H.; Kim, D.E. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy 2017, 13, 248–263.
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005.
- Song, C.; Mitter, S.K.; Qi, X.; Beli, E.; Rao, H.V.; Ding, J.; Ip, C.S.; Gu, H.; Akin, D.; Dunn, W.A., Jr.; et al. Oxidative stress-mediated NFkappaB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS ONE 2017, 12, e0171940.
- Lee, S.Y.; Oh, J.S.; Rho, J.H.; Jeong, N.Y.; Kwon, Y.H.; Jeong, W.J.; Ryu, W.Y.; Ahn, H.B.; Park, W.C.; Rho, S.H.; et al. Retinal pigment epithelial cells undergoing mitotic catastrophe are vulnerable to autophagy inhibition. Cell Death Dis. 2014, 5, e1303.
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510.
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953.
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693.
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518.
- Feng, Y.; Liang, J.; Zhai, Y.; Sun, J.; Wang, J.; She, X.; Gu, Q.; Liu, Y.; Zhu, H.; Luo, X.; et al. Autophagy activated by SIRT6 regulates Abeta induced inflammatory response in RPEs. Biochem. Biophys. Res. Commun. 2018, 496, 1148–1154.
- Di Rita, A.; Peschiaroli, A.; Pasquale, D.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755.
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564.
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221.
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080.
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002.
- Diot, A.; Morten, K.; Poulton, J. Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 2016, 27, 381–395.
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol. Vis. 2008, 14, 644–651.
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 2010, 31, 2002–2010.
- Gouras, P.; Ivert, L.; Neuringer, M.; Nagasaki, T. Mitochondrial elongation in the macular RPE of aging monkeys, evidence of metabolic stress. Graefe’s Arch. Clin. Exp. Ophthalmol. 2016, 254, 1221–1227.
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579.
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538.
- Liu, Q.; Greimann, J.C.; Lima, C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 2006, 127, 1223–1237.
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788.
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403.
- Klingeborn, M.; Stamer, W.D.; Bowes Rickman, C. Polarized Exosome Release from the Retinal Pigmented Epithelium. Adv. Exp. Med. Biol. 2018, 1074, 539–544.
- Hageman, G.S.; Mullins, R.F.; Russell, S.R.; Johnson, L.V.; Anderson, D.H. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999, 13, 477–484.
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846.
- Zhang, W.; Ma, Y.; Zhang, Y.; Yang, J.; He, G.; Chen, S. Photo-Oxidative Blue-Light Stimulation in Retinal Pigment Epithelium Cells Promotes Exosome Secretion and Increases the Activity of the NLRP3 Inflammasome. Curr. Eye Res. 2018, 1–9.
- Biasutto, L.; Chiechi, A.; Couch, R.; Liotta, L.A.; Espina, V. Retinal pigment epithelium (RPE) exosomes contain signaling phosphoproteins affected by oxidative stress. Exp. Cell Res. 2013, 319, 2113–2123.
- Tong, Y.; Zhou, Y.L.; Wang, Y.X.; Zhao, P.Q.; Wang, Z.Y. Retinal pigment epithelium cell-derived exosomes: Possible relevance to CNV in wet-age related macular degeneration. Med. Hypotheses 2016, 97, 98–101.
- Kang, G.Y.; Bang, J.Y.; Choi, A.J.; Yoon, J.; Lee, W.C.; Choi, S.; Yoon, S.; Kim, H.C.; Baek, J.H.; Park, H.S.; et al. Exosomal proteins in the aqueous humor as novel biomarkers in patients with neovascular age-related macular degeneration. J. Proteome Res. 2014, 13, 581–595.
- Elshelmani, H.; Rani, S. Exosomal MicroRNA Discovery in Age-Related Macular Degeneration. Methods Mol. Biol. 2017, 1509, 93–113.
- Feeney-Burns, L.; Eldred, G.E. The fate of the phagosome: Conversion to ‘age pigment’ and impact in human retinal pigment epithelium. Trans. Ophthalmol. Soc. UK 1983, 103 Pt 4, 416–421.
- Bosch, E.; Horwitz, J.; Bok, D. Phagocytosis of outer segments by retinal pigment epithelium: Phagosome-lysosome interaction. J. Histochem. Cytochem. 1993, 41, 253–263.
- Nandrot, E.F.; Kim, Y.; Brodie, S.E.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J. Exp. Med. 2004, 200, 1539–1545.
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15.
- D’Cruz, P.M.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; LaVail, M.M.; Vollrath, D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum. Mol. Genet. 2000, 9, 645–651.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
03 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No