Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Johanna Kusnick | + 4059 word(s) | 4059 | 2022-02-23 04:32:28 | | | |
| 2 | Rita Xu | Meta information modification | 4059 | 2022-03-03 02:29:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kusnick, J. Molecular Basis of Ferroptosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/20107 (accessed on 26 June 2026).
Kusnick J. Molecular Basis of Ferroptosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/20107. Accessed June 26, 2026.
Kusnick, Johanna. "Molecular Basis of Ferroptosis" Encyclopedia, https://encyclopedia.pub/entry/20107 (accessed June 26, 2026).
Kusnick, J. (2022, March 02). Molecular Basis of Ferroptosis. In Encyclopedia. https://encyclopedia.pub/entry/20107
Kusnick, Johanna. "Molecular Basis of Ferroptosis." Encyclopedia. Web. 02 March, 2022.
Copy Citation
Ferroptosis is a recently recognized iron-dependent form of non-apoptotic regulated cell death (RCD) characterized by lipid peroxide accumulation to lethal levels. Cancer cells, which show an increased iron dependency to enable rapid growth, seem vulnerable to ferroptosis. There is also increasing evidence that ferroptosis might be immunogenic and therefore could synergize with immunotherapies. Hepatocellular carcinoma (HCC) is the most common primary liver tumor with a low survival rate due to frequent recurrence and limited efficacy of conventional chemotherapies, illustrating the urgent need for novel drug approaches or combinatorial strategies. Immunotherapy is a new treatment approach for advanced HCC patients.
ferroptosis
immunotherapy
HCC
cancer
cell death
1. Introduction
Cell death is crucial for normal development and homeostasis throughout an organism’s lifetime [1]. Various forms of cell death have been identified, each having its respective modalities and features. Ferroptosis is one of the more recently described forms of non-apoptotic cell death. The term ferroptosis was first mentioned in 2012 by Dixon as a newly identified and unique iron-dependent cell death [2]. Ferroptotic cells display cytological changes that differ from morphological, biochemical, and genetic characteristics of other cell death forms, including decreased mitochondria cristae and ruptured mitochondrial membranes [3][4][5]. Compared to apoptosis, ferroptosis lacks rupture or blebbing of the plasma membrane, chromatin condensation, or rounding of the cell, and the execution of ferroptosis is not known to require specific pro-death protein expression [6]. As implied in its name, ferroptotic cell death is defined by the requirement of iron. Caused by excessive peroxidation of membrane phospholipids and production of reactive oxygen species (ROS), the plasma membrane loses its selective permeability, ultimately resulting in cell death. Regulated by multiple layers of metabolic signaling pathways including iron metabolism, lipid metabolism, and mitochondrial function, ferroptotic cell death is a complex process that will be further discussed in more detail. There is recent evidence suggesting that ferroptotic cell death plays an important role in mediating a wide variety of cellular processes in diseases, including immune activation. Whether ferroptotic cancer cells are immunogenic is currently unclear, but targeting ferroptosis in immunotherapeutic approaches is considered as a promising strategy.
Primary liver cancer is one of the world’s most common causes of cancer-related death and represents a serious health and economic burden [7][8]. Hepatocellular carcinoma (HCC) is the most common primary liver tumor, comprising around 75% of all liver cancer cases and typically developing in the context of liver fibrosis or cirrhosis [9]. In patients with advanced cirrhosis and HCC, liver transplant often remains the only curative treatment option [10]. Classic chemotherapies such as cisplatin are not regularly used in HCC due to the rapid development of chemoresistance [11], toxicity to various organs, and limited efficacy [12]. The first effective systemic therapy in advanced HCC was achieved using multi-kinase inhibitors, such as sorafenib, lenvatinib, carbozantinib, or regorafenib, but all of these substances provide only a rather small improvement of overall patient survival [13][14][15]. Treatment options are usually limited, as surgical resection or organ transplantation is feasible only in a fraction of patients and systemic therapies are not curative [16].
Antitumor immunotherapy has emerged as standard therapy for cancer treatment for many tumors (including HCC), and great progress has been achieved in the past few decades. The most successful immunotherapy to date is the modulation of immune checkpoint pathways, e.g., by targeting the T-cell inhibitory molecules programmed cell death-1 protein (PD-1) and programmed death-ligand 1 (PDL1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) that contribute to the tumor’s escape from cytotoxic T-cell responses [17][18]. Recently, a phase III trial reported that combination of atezolizumab (anti-PD-L1 antibody) and bevacizumab (anti-vascular endothelial growth factor receptor (VEGFR)) resulted in increased overall survival of patients with advanced HCC in comparison to sorafenib, making atezolizumab/bevacizumab the new first-line treatment for unresectable HCC [19]. Two other PD-1 inhibitors (nivolumab and pembrolizumab) have already been approved as a second-line monotherapy in advanced HCC in the USA, but the response rates are still low (15–20%) [20][21]. It has become increasingly clear that the efficacy of cancer immunotherapy depends on several factors including the potential to induce an anti-tumor immune response to overcome resistance. Immunogenic cell death (ICD) is seen as a promising concept in achieving strong and long-lasting anti-cancer immunity by not only killing malignant cells but also activating the immune system [22], which has the potential to synergize with immune checkpoint blockade. Despite ongoing research, less is known about regulators of ferroptosis and its role in immune activation, but there is some evidence that ferroptotic cell death might be immunogenic. These considerations support the concept to develop ferroptosis-based approaches to enhance response to immunotherapy.
2. Molecular Basis of Ferroptosis
Dixon et al. [2] first proposed ferroptosis in 2012 as a recently recognized non-apoptotic form of cell death. The knowledge about various cell death modalities and especially their understanding of ferroptosis is incomplete. Further research is necessary in order to understand why certain stimuli trigger ferroptosis instead of apoptosis. However, it is clear that ferroptosis describes an oxidative, iron-dependent process characterized by massive lipid peroxidation-mediated membrane damage and accumulation of ROS to toxic levels [1][5]. Its morphology differs from other known cell death types, such as apoptosis, necrosis, autophagy, necroptosis, or pyroptosis, as ferroptotic cells have characteristic mitochondrial atrophy accompanied by a reduction or disappearance of mitochondrial cristae [4][23]. Previous studies concur that ferroptosis mostly resembles necrosis-like morphological changes but depends mainly on iron signaling [24]. The two main biochemical characteristics are lipid peroxidation and iron accumulation, which will be detailed separately in the following sections.
2.1. Lipid Peroxidation and Antioxidant Defense
The peroxidation of polyunsaturated fatty acids (PUFAs) in phospholipids and their incorporation into the cell membrane are the main characteristics of ferroptosis (Figure 1) [25]. Catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4), PUFAs are incorporated into phospholipids to form polyunsaturated fatty acid-containing phospholipids (PUFA-PLs). Free PUFAs become a radical target after incorporation into the plasma membrane by ACSL4. Plasma membrane PUFAs in turn are vulnerable to free radical-initiated oxidation and trigger additionally self-amplified Fenton reaction, leading to destruction of the plasma membrane lipid bilayer and affecting membrane function [26]. The exact details remain unclear, but the peroxidation of phospholipid PUFAs is likely catalyzed by lipoxygenase (LOX) family enzymes [27]. There is recent evidence that the amount of PUFAs at the plasma membrane has an influence on the response to ferroptosis: n-3, but also, remarkably, n-6-long chain PUFAs showed cytotoxic effects proportional to the number of double bonds [28]. However, the antioxidant enzyme GPX4 is able to directly reduce phospholipid hydroperoxide to hydroxyphospholipid, which hinders the production of intracellular lipid hydroxyl radicals (Figure 1a). Glutathione (GSH) is considered as an essential cofactor for GPX4, as it can donate reducing equivalents for GPX4 opposing conversion of lipid hydroperoxides (L-OOH) to non-toxic lipid alcohols (L-OH) [29]. GSH synthesis is dependent upon cystine import into the cells by the cysteine/glutamate transporter (also known as system xc−, Figure 1b) which consists of two subunits, solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) [30]. The imported cystine can be oxidized to cysteine, which is then used to synthesize GSH required for GPX4 activity as a reducing cofactor. The GSH-GPX4 antioxidant system has an important role in protecting cells from ferroptosis as its activity determines the extent of ferroptosis by adjusting ROS accumulation and oxidative stress [31]. Pharmacological inhibition of the upstream regulator system xc− or the downstream effector GPX4 of GSH induces ferroptosis. The same can be achieved through depletion of system xc− (encoded by the SLC7A11 gene) or GPX4 genetic depletion [32].
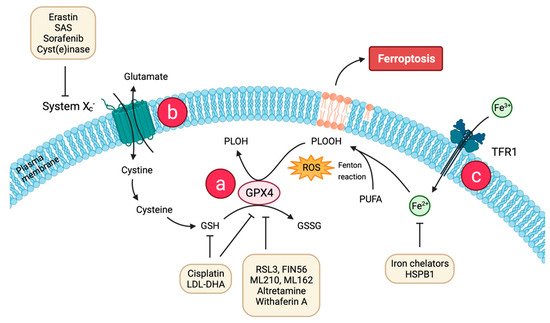
Figure 1. Molecular basis of ferroptosis and strategies for therapeutic induction of ferroptosis. A key cause of ferroptosis is the lipid radical chain reaction leading to excessive production and failure of elimination of phospholipid hydroperoxides (PLOOH). Two major mechanisms mediated by (a) glutathione peroxidase 4 (GPX4) and (b) system xc− serve to prevent PLOOH accumulation. System xc− mediates the uptake of cystine for the production of cysteine and glutathione (GSH). GPX4 converts GSH to glutathione disulfide (GSSG), resulting in reduced PLOOH and inhibition of ferroptosis. (c) Extracellular Fe3+ enters the cell through transferrin receptor 1 (TFR1) on the cell membrane, is reduced to Fe2+, and is combined with reactive oxygen species (ROS) to participate in lethal iron-dependent peroxidation of polyunsaturated fatty acids (PUFAs), and finally induces ferroptosis. Multiple ferroptosis-inducing agents (system xc−, GPX4 inhibitors, nanoparticles) hold great promise for cancer therapy. Created with BioRender. Available online: https://biorender.com (accessed on 10 February 2022). Abbreviations: SAS: sulfasalazine, LDL-DHA: ow-density lipoprotein docosahexaenoic acid, RSL3: Ras-selective lethal small molecule 3, FIN56: N2,N7-dicyclohexyl-9-(hydroxyimino)-9H-fluorene-2,7-disulfonamide, ML210:α-[(2-chloroacetyl)(3-chloro-4-methoxyphenyl)amino]-N-(2-phenylethyl)-2-thio-pheneacetamide, ML162:α-[(2-chloroacetyl)(3-chloro-4-methoxy-phenyl)amino]-N-(2-phenylethyl)-2-thiophene-acetamide, HSPB1: heat shock protein beta-1.
2.2. Iron Accumulation
As an essential trace element, iron is necessary to maintain cell metabolism [33]. Extracellular iron is internalized into the cell bound to transferrin (TF) as a TF-Fe3+ complex (Figure 1c). Taken up by the transferrin receptor 1 (TFR1) at the plasma membrane through clathrin-mediated endocytosis, it is released into the cytosol as free Fe2+ [34]. During ferroptosis, Fe2+ participates in Fenton chemistry to generate hydroxyl radicals und initiates lipid peroxidation, thus leading to propagation of damage throughout the membrane as already discussed [31].
Therefore, elevated levels of iron and iron derivates (heme or iron-sulfur clusters) can increase the vulnerability to ferroptosis, which plays an important role in tumor cells due to their extraordinary growth. Because of this increased susceptibility of cancer cells, ferroptosis might be a promising tool for anti-cancer treatment. Moreover, several proteins and genes that are involved in iron metabolism have been shown to modulate sensitivity to ferroptosis by changing cellular iron contents [35]. This emphasizes the association of cell death pathways with cell metabolism. Iron dependency also becomes apparent through blocking of TF-mediated iron import or recycling of iron-containing storage proteins, which leads to attenuation of ferroptosis [36]. Previous findings revealed involvement of other cell death types, contributing to ferroptosis and cell death fate: autophagic cell death requires the iron-carrier TF and the amino acid glutamine to trigger death [37]. Gao et al. demonstrated that ferroptosis is a form of autophagic cell death known as ferrotinophagy when autophagy is activated upon ferroptotic cell death induction. Autophagy is activated to degrade cellular ferritin and thereby increases iron levels and sensitizes to ferroptosis. The concentration of free ferrous ions in the cytoplasm determines whether it acts as a beneficial cofactor or as a toxic-free radical catalyst in the cell [38]. Growing evidence suggests that the autophagic machinery, at least under certain conditions and connected to human diseases, contributes to ferroptotic cell death and plays a dual role in tumorigenesis and, with it, cancer therapy [39].
Non-canonical ferroptosis induction by increasing intracellular Fe2+ refers to increasing iron intake through TFR1 expression or decreased expression of iron transporter, or excessive activation of heme oxygenase 1 (HMOX1) [40]. Heme metabolism influences iron contents, as it is a major source of dietary iron in mammals, derived primarily from hemoglobin and myoglobin. It plays an essential role in the liver, which is the principle organ that responds to changes in systemic iron signals in order to maintain body iron homeostasis [41]. As a central metabolic and signaling molecule, it is involved in diverse transcription and cell signaling processes, e.g., heme inhibits the proteasome, and binding to immunoglobulins [42][43]. HMOX1 catalyzes the catabolism of heme, which raises intracellular ferrous iron in the cytosol and is, therefore, crucial for maintaining cellular redox homeostasis. HMOX1 knockout mice showed iron accumulation in liver and kidney, and HMOX1 deficiency in humans is connected to several abnormalities, indicating an important role of HMOX1 in human health [44].
2.3. Regulatory Pathways
As described previously, ferroptosis is a regulated form of cell death driven by accumulation of membrane lipid peroxides from iron overload. Various genetic changes were considered important to contribute to this dynamic type of cell death. The major drivers of ferroptosis “decide” over life or death in response to ferroptotic stimuli. A predictive biomarker for monitoring ferroptosis appears to be the enzyme ACSL4 that is involved in fatty acid metabolism, contributing to accumulation of lipid intermediates [45]. Upregulation of ACSL4 enhances PUFAs content in phospholipids, which are susceptible to oxidation reactions. Expression levels of ACSL4 correlate with ferroptosis sensitivity—this was demonstrated by suppression of ACSL4 expression by RNA interference (RNAi) in HepG2 and HL60 cells, leading to an increase of ferroptosis resistance, while its overexpression restores ferroptosis sensitization [46]. As with factors involved in fatty acid metabolism, ferroptosis can also be regulated via signal transduction pathways of iron metabolism. Decreasing iron utilization may increase the sensitivity to ferroptosis whereas intracellular iron pools seem to promote ferroptosis [47]. Various genes and proteins related to iron metabolisms are generally upregulated during ferroptosis, including the iron responsive element binding protein 2 (IREB2). IREB2 is considered the core regulator of iron metabolism binding iron-responsive elements in the mRNA of target genes including the TF receptors and knockdown results in resistance to ferroptosis induction [2]. Additionally, overexpression of iron-storage proteins such as ferritin are supposed to play a wide anti-ferroptotic role [48].
GPX4 is the major lipid hydroperoxide (LOOH)-neutralizing enzyme, which in turn represents a target for several regulating proteins. Erastin and Ras-selective lethal small molecule 3 (RSL3) both inactivate GPX4 and are proposed as efficient ferroptosis inducers. Erastin does so indirectly by inhibiting cystine import, which then lacks as an essential cellular building block of GSH. Erastin is also a potential activator of mitochondrial voltage-dependent anion channel 2/3 (VDAC2/3), highlighting the participation of mitochondria dysfunction in erastin-induced ferroptosis [49]. RSL3 directly binds the active site of GPX4 and inhibits the phospholipid peroxidase activity [50].
Research on the mechanism of ferroptosis also required the identification of ferroptosis inhibitors in order to study the role of ferroptosis in vivo and discuss potential pharmacological approaches aiming to subvert lipid peroxidation. Ferrostatin-1 (Fer-1), liproxstatin-1, vitamin E, and zileuton have been identified and suppress ferroptosis as lipoxygenase inhibitors, preventing the propagation of oxidative damage within the membrane [51][52]. Other than that, iron chelators such as deferoxamine inhibit the accumulation of iron [2]. A full understanding of the regulatory network including ferroptosis inhibitors might provide potential health benefits to prevent symptoms of diseases that are related to ferroptosis [53].
In summary, many potent biomarker candidates have already been identified, which could be useful to further study the fundamental basis of lipid metabolism in ferroptosis and to define the interplay of involved regulators. However, further functional studies are required to unravel the critical role of certain biomarkers in order to find applications in ferroptosis-based cancer therapy.
2.4. Immunogenic Features of Ferroptosis (in Cancer Cells)
There is rising evidence supporting the notion that ferroptosis can play a significant role in mediating various functions during immune responses and immunotherapies. For early ferroptotic cancer cells, it has been proven that they release adenosine triphosphate (ATP) and high mobility group box 1 (HMGB1), which can be recognized as damage-associated molecular patterns (DAMPs) by certain immune cells, triggering an inflammatory response [54]. At present, the most effective systemic therapy in advanced HCC is the combinational approach of atezolizumab and bevacizumab [19]. The response rate is largely dependent on the immune status of the tumor: inflamed (“hot”) tumors with high infiltration of tumor-reactive T cells respond significantly better to checkpoint inhibitors than “cold” or “excluded” tumors that are poorly infiltrated by immune cells including antigen-presenting cells (APCs) [55][56]. Immune checkpoint inhibitors (such as PD-1 or CTLA4) mainly act by activating effective anti-cancer immune responses driven by cytotoxic T cells. Ferroptosis in cancer cells can be induced by CD8+ T cells that release INF-γ, which leads to system xc− downregulation and consequently lipid peroxidation [57].
Compared to other forms of cell death, ferroptosis is considered an immune-activating process. Although further research is still needed to clarify the interaction between ferroptotic cell death and the immune system, there is some preliminary evidence showing that ferroptosis plays a direct role for the immune system. This is not surprising since iron metabolism and lipid peroxidation were linked to regulation of immune functions, even before ferroptosis was discovered. Further determination of specific immune signals associated with ferroptosis would be useful in order to enhance the immune effect in tumor immunotherapy.
Typically for RCD, DAMPs are released as immune modulators, stimulating immune cells (e.g., macrophages or monocytes) (Figure 2) [58]. These are endogenous molecules that bind to pattern recognition receptors (PRRs), thus activating the production of immune stimulants, including a variety of cytokines and chemokines. A typical DAMP, which is released by ferroptotic cells in an autophagy-dependent manner, is the nuclear transcription factor HMGB1 able to bind chromosomal DNA involved in DNA recombination and repair processes [59]. It was shown to be actively secreted by innate immune cells in response to microbial infections but can also be passively released by somatic cells undergoing cytoplasmic membrane destruction during cell death [60]. HMGB1 acetylation induced by histone deacetylase inhibition is the trigger for HMGB1 release during ferroptosis [61]. Moreover, another study shows that HMGB1 is an essential factor for cancer cell immunity and plays paradoxical roles in tumor development by promoting both cell survival and death [62]. For a more complete investigation of the full immunogenic potential of this cell death pathway, it is important to discover the full repertoire of DAMPs released during ferroptosis. While the role of HMGB1 and ATP release has already been clarified in apoptosis and necroptosis [63], its involvement in ferroptotic cell death processes has remained elusive until now. Recent research highlights the involvement in the regulation of TFR mRNA levels and p38 phosphorylation in the rat sarcoma (RAS)-JNK/p38 pathway [64]. Additionally, Ye et al. found that knockdown of HMGB1 reduces erastin-mediated ferroptosis and the connected ROS generation [64]. Hence, HMGB1 is a critical regulator of ferroptosis.
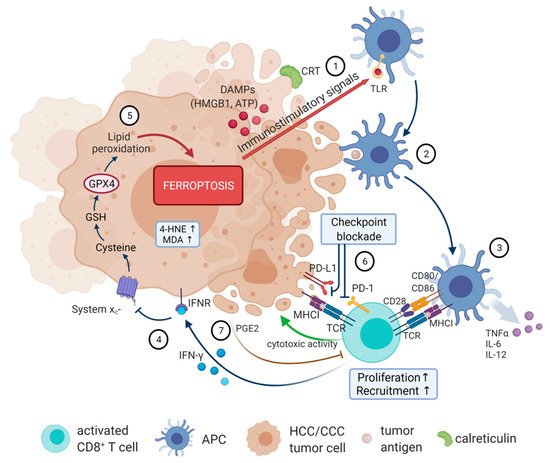
Figure 2. The immunogenic features of ferroptotic cancer cells. Ferroptotic cells release DAMPs, including HMGB1, ATP, and CRT (1), that act as immune modulators inducing maturation of antigen-loaded DCs (2). Activated DC release pro-inflammatory cytokines and present TAA to T cells (3). Activated CD8+ T cells release IFN-γ, activating INFR, which, in turn, downregulates expression of glutamate-cystine antiporter system xc− (4), and consequently promotes tumor cell lipid peroxidation and ferroptosis (5). Immune-checkpoint blockade of PD-1 enhances antitumor responses against cancers (6). Simultaneously, ferroptotic cancer cells release PGE2 that suppresses cytotoxic T cell activity (7). Created with BioRender. Available online: https://biorender.com (accessed on 10 February 2022). Abbreviations: APC: antigen-presenting cell; ATP: adenosine triphosphate; CD: cluster of differentiation; CRT: calreticulin; DAMPs: damage-associated molecular patterns; DC: dendritic cell; GPX4: glutathione peroxidase 4; GSH: glutathione; HMGB1: high mobility group box 1; 4-HNE: 4-hydroxynonenal; IFN-γ: interferon-γ; IFNR: interferon-receptor; IL: interleukin; MDA: malondialdehyde; MHC: major histocompatibility complex; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; PGE2: prostaglandin E2; TAA: tumor associated antigen; TCR: T-cell receptor; TLR: toll-like receptor; TNFα: tumor necrosis factor α.
Furthermore, other immune signals can be intermediate or final products of lipid peroxidation such as 4-hydroxynonenal (4-HNE) or malondialdehyde (MDA), driving inflammation by activating macrophages to produce pro-inflammatory cytokines [65]. They can trigger inflammation partly through activating TLR4 signaling [66]. However, thus far, there is no direct evidence that these toxic aldehydes are alone sufficient to induce ferroptosis. Taken together, despite the mechanisms reviewed here, current evidence is insufficient for a complete understanding of the relation between ferroptosis and the immune system. A more comprehensive understanding of DAMP release mechanisms and their regulation could enable development of potential new drugs for therapeutic interventions in cancer.
2.5. Immune Response to DAMPs Released by Ferroptotic Cells
Until now, two ways for ferroptotic cells to influence inflammatory responses in immune cells have been characterized. On the one hand, the release of DAMPs by non-immune ferroptotic cells attract APCs and other immune cells. Secretion of DAMPs attracts and stimulates dendritic cells (DCs) for tumor-associated antigen (TAA) uptake and processing followed by MHC class I-restricted cross-presentation of these TAAs, which leads to the priming of T cells and clonal expansion of cancer-specific cytotoxic T lymphocytes (Figure 2) [67]. For example, HMGB1 is able to trigger an inflammatory response in peripheral macrophages via activation of the NF-κB pathway [59]. Functioning as an adjuvant, HMGB1 contributes to activation of the innate and adaptive immune systems. Targeting HMGB1 release using neutralizing anti-HMGB1 antibodies limits inflammatory response in macrophages during ferroptosis [59]. On the other hand, ferroptosis in immune cells themselves might compromise immune responses. It has been shown that non-apoptotic cell death, including ferroptosis, is more likely to cause inflammation than apoptosis but little is known about the recognition and clearance mechanisms for cells dying through non-apoptotic cell death pathways. Phagocytic clearance of dead cells plays a vital role in immune homeostasis, especially during cancer therapy, and the exact mechanistic of ferroptotic cell engulfment remains unclear. However, Luo et al. have demonstrated that different types of macrophages readily engulfed ferroptotic cells through a mechanism different from the engulfment of apoptotic cells. Ferroptotic cells can produce oxygenated phosphatidylethanolamines on the outer plasma membrane that can be recognized by TLR2 on macrophages, leading to clearance of ferroptotic cells [68]. Moreover, macrophages can be classified as pro-inflammatory or anti-inflammatory pro-carcinogenic macrophages, both functioning in eliminating pathogens in order to maintain immune homeostasis. Environmental signals decide on pro- and anti-inflammatory polarization, whereas an imbalance contributes to various diseases and tumor development [69], and previous studies indicated a relationship between macrophage polarization and ferroptosis. Dai et al. showed that ferroptotic cancer cells cause autophagy-dependent protein release, triggering pro-carcinogenic macrophage polarization via activation of several signaling pathways, which could restrict anti-tumor immunity [70]. Moreover, macrophage subgroups differ concerning their sensitivities to ferroptosis induction, with pro-inflammatory ones being more susceptible [71]. Regarding the adaptive immune system, T cells can also induce ferroptosis in tumor cells. It has been reported that CD8+ T cells release cytokines (e.g., TNF or IFN-γ), which significantly downregulate expression of system xc− components driving tumor cells into ferroptosis due to reduced cysteine uptake [57]. This might provide a direction for further bridging the gap between ferroptosis and immunotherapy in the treatment of malignant cancers.
Despite system xc−, the other key regulator of ferroptosis, GPX4, was also reported to play an essential role in the expansion of both antigen-specific CD4+ and CD8+ T cells [72]. A lack of GPX4 leads to ferroptotic cell death induction with an accumulation of lipid peroxides in T cells, thereby preventing their immune response to infection. Moreover, it was recently found that ferroptosis-resistant CD8+ T cells overexpressing GPX4 have normal immune functions, but deletion of ACSL4 impairs their immune response [73]. GPX4 also plays a pivotal role in preventing ferroptosis after T-cell activation by only expanding GPX4-expressing CD4+ and CD8+ T cells [72]. Additionally, the induction of ferroptosis in cancer cells has been shown to be associated with increased release of prostaglandin E2 (PGE2), an immunosuppressive agent directly suppressing cytotoxic T-cell activity (Figure 2 (7)). Therefore, PGE2 promotes the proliferation of tumor cells and plays a central role in impairing anti-tumor immunity, which could be a barrier to the induction of a potent immune response [74]. Thus, understanding the molecular mechanisms underlying the immune response of ferroptotic cell death would help guide the design of new immune-related treatments.
To do so, a more detailed investigation of the TME including activated immune cells that secrete proinflammatory chemokines and cytokines is necessary. Recent studies confirmed the role of ferroptotic cancer cells regulating tumor immunity in different ways. As other cell death types, ferroptotic cells release “find me” signals that attract APCs and other immune cells, leading to the engulfment by macrophages [75]. During maturation and cross-presentation by DCs, ferroptotic cancer cells interact further with the immune system through TLR4 [76].
In a very recent integrative study, a ferroptosis scoring model based on ferroptosis-related genes and their association with immune infiltration in cancer was constructed [77]. For this purpose, 30 genes were classified into ferroptosis driver and suppressor groups, and transcription factors and therapeutic agents targeting ferroptosis regulators in cancer were determined and validated. This score can be a potential biomarker of the immune response in the TME and the therapeutic response to immunotherapy. The described dual effects of ferroptosis on tumor immunity, and with it, patient’s prognosis score, differ from person to person, but the results showed that the ferroptosis score, as an independent prognostic factor, is feasible for predicting the prognosis of different cancers and immunotherapy efficacy. Supporting previously obtained results, high ferroptosis scores had higher immune cell infiltration scores due to CD8+ T cell and T-helper cell activity [57]. This highlights the importance of the TME that should be considered in the design of therapeutic compounds and confirms the powerful combinatorial use of T cell-mediated ferroptosis induction with checkpoint blockade [78]. Despite some limitations of this study (more datasets for verification and combinatorial approaches would be needed), this ferroptosis score might have potential to help improve the survival rate of patients with HCC and predict tumor progression earlier as prognostic markers that currently find application in clinic.
References
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209.
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34.
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13.
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global Trends in Incidence Rates of Primary Adult Liver Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 171.
- Lurje, I.; Czigany, Z.; Bednarsch, J.; Roderburg, C.; Isfort, P.; Neumann, U.P.; Lurje, G. Treatment Strategies for Hepatocellular Carcinoma (-) a Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 1465.
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883.
- Kumari, R.; Sahu, M.K.; Tripathy, A.; Uthansingh, K.; Behera, M. Hepatocellular carcinoma treatment: Hurdles, advances and prospects. Hepat. Oncol. 2018, 5, HEP08.
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390.
- Ikeda, M.; Okusaka, T.; Mitsunaga, S.; Ueno, H.; Tamai, T.; Suzuki, T.; Hayato, S.; Kadowaki, T.; Okita, K.; Kumada, H. Safety and Pharmacokinetics of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 1385–1394.
- Abou-Alfa, G.K.; Schwartz, L.; Ricci, S.; Amadori, D.; Santoro, A.; Figer, A.; De Greve, J.; Douillard, J.Y.; Lathia, C.; Schwartz, B.; et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006, 24, 4293–4300.
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179.
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429.
- Schmidt, N.; Flecken, T.; Thimme, R. Tumor-associated antigen specific CD8(+) T cells in hepatocellular carcinoma—a promising target for immunotherapy. Oncoimmunology 2014, 3, e954919.
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905.
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2021, 23, 77–90.
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202.
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337.
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front. Neurosci. 2018, 12, 214.
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147.
- Wang, L.; Chen, X.; Yan, C. Ferroptosis: An emerging therapeutic opportunity for cancer. Genes Dis. 2020, 9, 334–346.
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90.
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975.
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715.
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248.
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.
- Xiaoxuan, W.; Zicheng, L.; Lijuan, M.; Haijie, Y. Ferroptosis and its emerging role in tumor. Biophys. Rep. 2021, 7, 280.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125.
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153.
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849.
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Lan, H.; Gao, Y.; Zhao, Z.; Mei, Z.; Wang, F. Ferroptosis: Redox Imbalance and Hematological Tumorigenesis. Front. Oncol. 2022, 12.
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435.
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12.
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226.
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125.
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol. Med. 2016, 22, 200–213.
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem. Biol. 2018, 13, 1996–2002.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98.
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343.
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643.
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308.
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176.
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci 2018, 4, 387–396.
- Sharma, A.; Flora, S.J.S. Positive and Negative Regulation of Ferroptosis and Its Role in Maintaining Metabolic and Redox Homeostasis. Oxid. Med. Cell Longev. 2021, 2021, 9074206.
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8.
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168.
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330.
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274.
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175.
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283.
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001.
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162.
- Tang, D.; Kang, R.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys Acta 2010, 1799, 131–140.
- Demuynck, R.; Efimova, I.; Naessens, F.; Krysko, D.V. Immunogenic ferroptosis and where to find it? J. Immunother. Cancer 2021, 9.
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 2019, 9, 730–739.
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199.
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242.
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72.
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989.
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566.
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083.
- Luo, L.; Wang, H.; Tian, W.; Zeng, J.; Huang, Y.; Luo, H. Targeting ferroptosis for cancer therapy: Iron metabolism and anticancer immunity. Am. J. Cancer Res. 2021, 11, 5508–5525.
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568.
- Drijvers, J.M.; Gillis, J.E.; Muijlwijk, T.; Nguyen, T.H.; Gaudiano, E.F.; Harris, I.S.; LaFleur, M.W.; Ringel, A.E.; Yao, C.H.; Kurmi, K.; et al. Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8(+) T Cells. Cancer Immunol. Res. 2021, 9, 184–199.
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28.
- Kloditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65.
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059.
- Tang, B.; Yan, R.; Zhu, J.; Cheng, S.; Kong, C.; Chen, W.; Fang, S.; Wang, Y.; Yang, Y.; Qiu, R.; et al. Integrative analysis of the molecular mechanisms, immunological features and immunotherapy response of ferroptosis regulators across 33 cancer types. Int. J. Biol. Sci. 2022, 18, 180–198.
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685.
More
Information
Subjects:
Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
745
Revisions:
2 times
(View History)
Update Date:
03 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No