Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tomas Simurda | + 1904 word(s) | 1904 | 2022-02-21 07:52:10 | | | |
| 2 | Jason Zhu | -52 word(s) | 1852 | 2022-03-02 03:16:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Simurda, T. Fibrinogen in Hypofibrinogenemia. Encyclopedia. Available online: https://encyclopedia.pub/entry/20046 (accessed on 21 July 2026).
Simurda T. Fibrinogen in Hypofibrinogenemia. Encyclopedia. Available at: https://encyclopedia.pub/entry/20046. Accessed July 21, 2026.
Simurda, Tomas. "Fibrinogen in Hypofibrinogenemia" Encyclopedia, https://encyclopedia.pub/entry/20046 (accessed July 21, 2026).
Simurda, T. (2022, March 01). Fibrinogen in Hypofibrinogenemia. In Encyclopedia. https://encyclopedia.pub/entry/20046
Simurda, Tomas. "Fibrinogen in Hypofibrinogenemia." Encyclopedia. Web. 01 March, 2022.
Copy Citation
Congenital fibrinogen disorders are diseases associated with a bleeding tendency; however, there are also reports of thrombotic events. Fibrinogen plays a role in the pathogenesis of thrombosis due to altered plasma concentrations or modifications to fibrinogen’s structural properties, which affect clot permeability, resistance to lysis, and its stiffness. Several distinct types of genetic change and pathogenetic mechanism have been described in patients with bleeding and a thrombotic phenotype, including mutations affecting synthesis or processing in three fibrinogen genes.
fibrinogen
hypofibrinogenemia
heterogeneity of phenotype
1. Fibrinogen’s Structure and Its Function
Fibrinogen is a 340 kDa glycoprotein that circulates in the blood [1]. The fibrinogen molecule is formed from a hexamer that consists of two copies of three nonidentical polypeptide chains (Aα, Bβ, and γ) [2]. Aα chains contain 610 residues, Bβ chains 461 residues, and the major γ chains consist of 411 residues. These polypeptide chains, Aα, Bβ, and γ, are connected by disulfide bridging [3]. Fibrinogen has a characteristic trinodal organization, consisting of a single central and two distal nodes [4]. The molecule has a 45 nm elongated structure with two outer D domains, which are connected to a central E domain by a coiled-coil segment [5]. Three independent genes encoding fibrinogen are clustered on chromosome 4 (region 4q31.3) [6]. Each chain of fibrinogen is encoded by a separate gene. The gene coding for the fibrinogen Aα chain (FGA) has a 7.6 kb size and consists of 6 exons, the Bβ chain gene (FGB) has 8 exons and occupies an 8.0 kb sized region, and the γ chain gene (FGG) encompasses an 8.5 kb region and consists of 10 exons [7]. Normal fibrinogen blood levels vary and are typically given as 1.8–4.2 g/L [8]. Fibrinogens belong to the acute-phase proteins whose levels increase in response to different stressful situations, such as tissue injury, inflammation, and the accompanying release of cytokines. Upregulation of fibrinogen expression is controlled by interleukin 6 (IL 6) and the glucocorticoid signaling pathways [9]. This causes a rapid increase in fibrinogen plasma levels after clotting events or bleeding, and in order to support wound healing [10]. Contrarily, transforming growth factor β (TGF–β), and cytokines IL4, IL10, and IL13 are negative regulators of transcription [9]. Fibrinogen is dominantly expressed in hepatocytes. However, extrahepatic production has been demonstrated in epithelial cells from the lungs, intestine, and cervix. For some years, biosynthesis of fibrinogen by megakaryocytes has been discussed, but it is widely believed that the fibrinogen present in alpha granules of thrombocytes originates primarily from plasma uptake [11].
2. Congenital Hypofibrinogenemia
Congenital hypofibrinogenemia is a quantitative fibrinogen disorder characterized by a proportional decrease in functional and antigenic fibrinogen levels [12][13]. On the basis of the level of fibrinogen concentration, hypofibrinogenemia is classified as severe, moderate, or mild. Certain hypofibrinogenemic variants result in liver disease, i.e., fibrinogen storage disease (FSD), which is caused by the intrahepatic accumulation of fibrin [14]. The classifications according to fibrinogen concentration levels are listed in Figure 1.
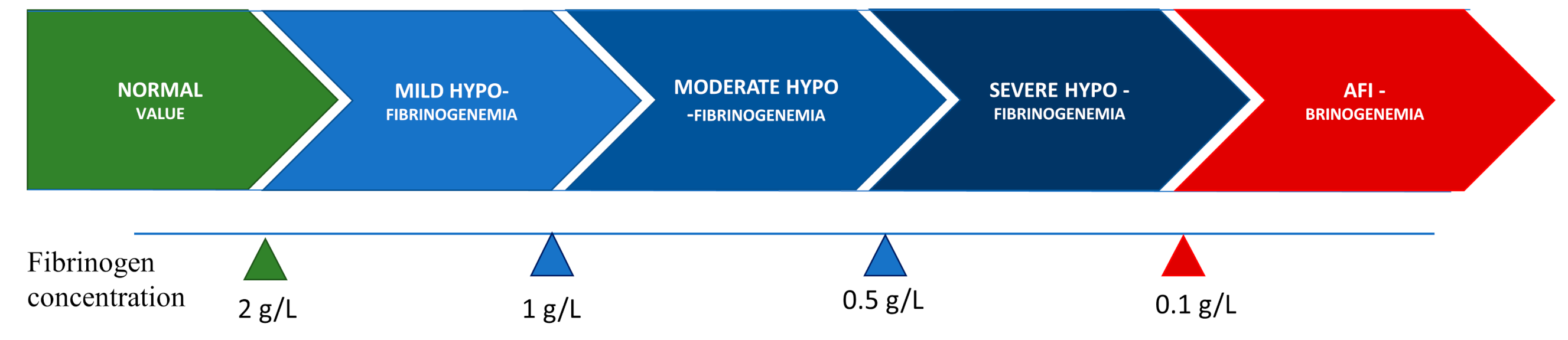
Figure 1. Classification of hypofibrinogenemia according to fibrinogen concentration.
Hypofibrinogenemia has diverse phenotypical variability, which can range from no manifestations to bleeding and/or thrombosis. Hypofibrinogenemic patients have a lower risk of bleeding than patients with afibrinogenemia [15]. Patients with hypofibrinogenemia are frequently asymptomatic and are diagnosed during routine laboratory testing, before invasive procedures, or in the setting of familial explorations. Hypofibrinogenemia can lead to a milder bleeding pattern, with surgery and trauma, with related bleeding significantly more common than spontaneous events (80% vs. 20%). The most typical bleeding symptoms are menorrhagia, hemorrhage from mucosal tracts, hemarthroses, hematomas, and GI bleeding [11]. Abnormalities of the fibrinogen molecule have been implicated in various adverse pregnancy outcomes, like placental abruption, postpartum hemorrhage, and spontaneous abortion, highlighting the role of fibrinogen in implantation and placentation [16]. Hypofibrinogenemic patients when exposed to other thrombotic risks factors, biological or acquired, they can also develop thrombosis in venous and/or arterial sites. Thus, clinical manifestations are not only the direct result of the fibrinogen level in circulation but also the result of additional personal, genetic, and environmental factors, such as smoking, use of oral contraceptives, immobilization, comorbid conditions, hormonal status, further thrombophilia and bleeding risk factors, amongst others.
3. Fibrinogen’s Role in the Pathophysiology of Thrombosis and Bleeding in Hypofibrinogenemia
Fibrinogen plays a crucial function in controlling bleeding after vascular injury by providing the support for platelet aggregation and the substrate for fibrin clotting [17]. Via the activated form of glycoprotein IIb/IIIa (known as integrin αIIbβ3), fibrinogen creates a scaffold for the aggregation of platelets. As a part of the rapid primary hemostatic response, thrombocyte aggregation via fibrinogen crosslinking contributes to the creation of an initial hemostatic barrier after blood vessel injury [18]. Afterwards thrombin is activated on surface of platelets and fibrinogen is converted into insoluble fibrin [19]. The last step in the blood clotting cascade necessary for hemostasis and thrombosis is the conversion of fibrinogen to fibrin [20]. Fibrinogen’s conversion to fibrin stops bleeding by providing the insoluble matrix of the blood clot [21]. As the fibrin is formed, the serine protease thrombin rapidly cleaves fibrinogen, releasing two fibrinopeptides, i.e., fibrinopeptide A (FpA, residues 1–16) and fibrinopeptide B (FpB, residues 1–14), from the N-terminal of the Aα and Bβ chains, respectively, and converts fibrinogen into fibrin monomers [22]. Fibrin monomers spontaneously polymerase, resulting in the formation of thicker fibers and an insoluble multistranded and branched fiber network that entangles platelets to form a blood clot, blocking the damaged blood vessel and preventing further bleeding [23][24].
Thus, the absence of normal fibrinogen leads to the disruption of said mechanisms and thus to bleeding complications (Figure 2). According to the literature data, the severity and pattern of clinical manifestations of bleeding are dependent on the fibrinogen levels [25]. With fibrinogen levels of around 1.0 g/L, patients with hypofibrinogenemia are usually asymptomatic. In theory, these levels are high enough to protect against bleeding and maintain pregnancy [26]. The bleeding phenotype usually develops in patients with fibrinogen levels lower than 1.0 g/L. A mean activity level of fibrinogen of at least 0.7 g/L prevents spontaneous hemorrhage [27].
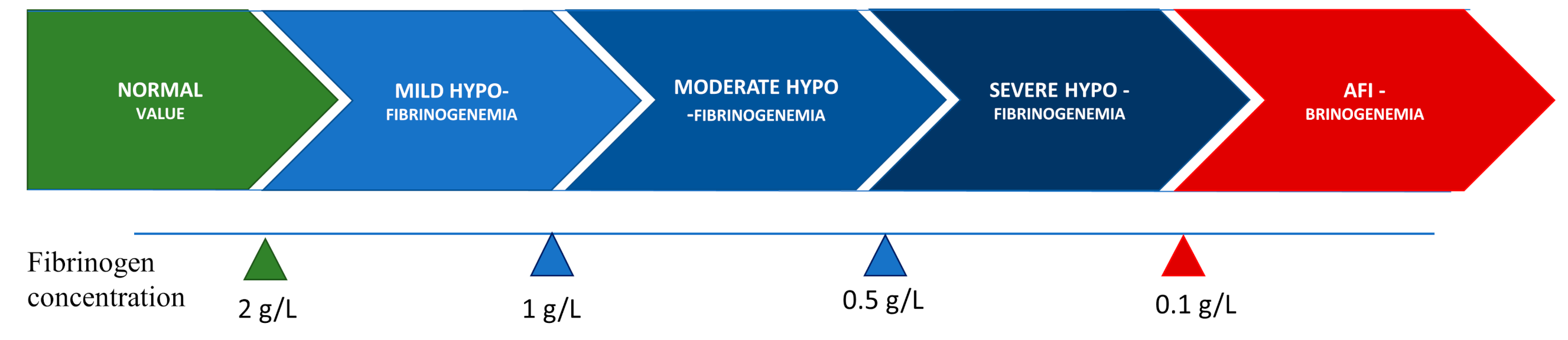
Figure 2. Pathogenesis of thrombosis and bleeding in association with low fibrinogen activity.
Fibrinogen mediates the modulation of coagulation and fibrinolysis through thrombin binding, conferring antithrombin activity, and through FXIII, plasminogen, antiplasmin, and tissue-type plasminogen activator (t-PA) [28]. Fibrinogen contributes to various pathological events including thrombosis due to decrease of plasma concentration of fibrinogen, its changed structural properties, or from the effect of polymorphisms on clot stiffness, permeability, and resistance to lysis (Figure 2) [10]. The reasons for the increased thrombotic risk in congenital fibrinogen disorders (CFD) are not entirely understood [29]. Thrombosis can develop in both arterial and venous sites. These events are less common in hypofibrinogenemic patients than in afibrinogenemic patients. The low level of circulating fibrinogen in hypofibrinogenemia is generally thought to be enough to lower the risk of developing thrombosis, which is more frequently described in afibrinogenemia. It is important to mention that low fibrinogen levels do not compensate for a hypercoagulable state [27].
One explanation for the increased risk of a thrombotic event in hypofibrinogenic patients is that circulating thrombin levels increase in association with fibrinogen deficiency. Thrombin is normally sequestered by the developing fibrin, thus reducing the amount of free thrombin in circulation. Free thrombin has the potential effect of platelet activation [29]. Fibrin deficiency reduces its antithrombin effect, causing an increase in circulating thrombin levels. Free thrombin, which is not trapped in the fibrin clot, activates platelets and stimulates the migration and proliferation of smooth muscle cells, leading to large and loose platelet thrombi [27]. Fibrin can also lower thrombin generation. Therefore, an absence or reduction of functional fibrinogen can be responsible for increased thrombin activity [29]. Soluble fibrinogen also competitively reduces platelet adhesion to immobilized fibrinogen. However, hemostasis in patients with quantitative fibrinogen disorders enables adequate thrombus formation. As a result of the lack of fibrin in clots, they become unstable and have tendency to embolize [30].
The clinical data are controversial as regards fibrinogen replacement therapy. It has been suggested that treatment with fibrinogen concentrates may be a risk factor for thrombosis. However, it should be noted that there is no clear evidence of a direct link between fibrinogen concentrate administration and the development of thrombosis. The pathogenesis at the basis of the paradoxical thrombotic tendency in patients with fibrinogen deficiency is likely multifactorial, depending on different exogeneous and endogenous risk factors, such as genetic thrombophilia, immobilization, pregnancy, surgery, or trauma. Thrombosis is described in more than one-third of hypofibrinogenemic patients after surgery, delivery, puerperium, and trauma. Thrombophilic mutations (i.e., protthombin G20210 and factor V Leiden) [19] have been reported in a small number of patients with CFD.
4. Review of Mutations Associated with Hypofibrinogenemia and Both Bleeding and Thrombotic Phenotype
Quantitative fibrinogen deficiencies are characterized by the concomitant reduction or absence of coagulant activity and immunoreactive proteins. Complete mutational screening of all three fibrinogen genes (FGA, FGB, FGG) is required for the molecular diagnosis of congenital fibrinogen disorders [31]. While many of the polymorphisms and minor alterations do not influence fibrinogen’s structure, function [32], or the multimeric form of fibrin, and are thus seen to be neutral, some of the more significant changes have been shown to influence fibrinogen’s function, structure, or both [33]. The spectrum of abnormal changes in molecular structure is broad, resulting into several subtypes of fibrinogen disorders with specific clinical and biological features [14].
Researchers distinguish two main classes of causative mutations: mutations producing abnormal protein chains, which are retained inside the cell; and null mutations with no protein production at all. Hypofibrinogenemia is generally caused by heterozygosity for these mutations.
Researchers searched for human fibrinogen variants associated with hypofibrinogenemia and both bleeding and thrombosis in the mutation database GFHT (French Study Group on Hemostasis and Thrombosis). In this database, new variants in all three genes, i.e., FGA, FGB, and FGG, for fibrinogen are regularly added. The GFHT database is available via the Study Group on Hemostasis and Thrombosis: http://www.geht.org/databaseang/fibrinogen, accessed on 6 October 2021, which lists all fibrinogen variants identified to date in patients with afi-, hypo-, dys-, and hypodysfibrinogenemia. As of 6 October 2021, the GFHT database lists 1215 molecular abnormalities associated with fibrinogen.
Researchers looked at all of the gene mutations in all three fibrinogen genes. Overall, in this database, researchers found 69 such mutations. Among them, 24 mutations resulted in afibrinogenemia, 15 in hypofibrinogenemia, 22 in dysfibrinogenemia, with the least common disorder being hypodysfibrinogenemia, which is caused by eight mutations. Most mutations were detected in the FGA gene. The lowest number of variants was found in the FGB gene.
On the basis of the GFHT database, a total of 15 mutations responsible for both a hypofibrinogenemia and a thrombotic and bleeding phenotypes were identified: three in FGA, five in FGB, and seven in FGG. Eight mutations were heterozygous, two homozygous, and five cases were the result of compound heterozygosity. Thirteen were localized in exons and two in introns. Most mutations were caused by genetic changes in exon 8 of FGG. The majority of patients with hypofibrinogenemia and mutations determining both the bleeding and thrombotic phenotype experienced venous thrombosis. The most common were pulmonary embolism, deep venous thrombosis, cerebral venous thrombosis, etc. Thrombotic events in the arterial circulation, such as myocardial infarction, ischemic stroke, peripheral arterial thrombosis, or arterial thrombosis at other sites, were less frequent. Thrombotic recurrences were mainly present in the venous territory. In addition, certain mutations were reported to be associated with at least with one miscarriage.
References
- Casini, A.; De Moerloose, P. Fibrinogen concentrates in hereditary fibrinogen disorders: Past, present, future. Haemophilia 2019, 26, 25–32.
- Cronje, H.T.; Nienaber-Rousseau, C.; Zandberg, L.; Chikowore, T.; Lange, Z.; van Zyl, T.; Pieters, M. Candidate gene analysis of the fibrinogen phenotype reveales the importance of polygenic co-regulation. Matrix Biol. 2016, 60, 16–26.
- Ridgway, H.J.; Brennan, S.O.; Loreth, R.M.; George, P.M. Fibrinogen Kaiserlautern (γ380 Lys to Asn) a new glycosylated fibrinogen variant with delayed polymerisation. Br. J. Haematol. 1997, 99, 562–569.
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 93, 11–30.
- Mosesson, M.W. Structure and function of fibrinogen and fibrin. J. Thromb. Haemost. 2005, 3, 1894–1904.
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclikova, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 111, 795–802.
- Tiscia, G.L.; Margaglione, M. Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. Int. J. Mol. Sci. 2018, 19, 1597.
- Simurda, T.; Asselta, R.; Zolkova, J.; Brunclikova, M.; Dobrotova, M.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Congenital Afibrinogenemia and Hypofibrinogenemia: Laboratory and Genetic Testing in Rare Bleeding Disorders with Life-Threatening Clinical Manifestations and Challenging Management. Diagnostics 2021, 11, 2140.
- Weasel, J.W.; Dempfle, C.E.H. Fibrinogen Structure and Function. Available online: https://oncohemakey.com/fibrinogen-structure-and-function (accessed on 7 September 2021).
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296.
- Asselta, R.; Duga, S.; Tenchini, M.L. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006, 4, 2115–2129.
- Simurda, T.; Caccia, S.; Asselta, R.; Zolkova, J.; Skornova, I.; Snahnicanova, Z.; Loderer, D.; Lasabova, Z.; Kubisz, P. Congenital hypofibrinogenemia associated with as novel heterozygous nonsense mutation in the globular C—Terminal domain of the γ—Chain (p.Glu275Stop). J. Thromb. Thrombolysis 2019, 50, 233–236.
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A Novel Nonsense Mutation in FGB (c.1421 G>A;p.Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605.
- Casini, A.; Undas, A.; Palla, R.; Thachil, J.; de Moerloose, P. Diagnosis and classification of congenital fibrinogen disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1887–1890.
- Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic variants in the FGB and FGG genes mapping in the beta and gamma nodules of the fibrinogen molecule in congenital quantitative fibrinogen disorders associated with a thrombotic phenotype. Int. J. Mol. Sci. 2020, 21, 4616.
- Miesbach, W.; Galanakis, D.; Scharrer, I. treatment of patients with dysfibrinogenemia and history of abortion during pregnancy. Blood Coagul. Fibrinolysis 2009, 20, 366–370.
- Asselta, R.; Platé, M.; Robusto, M.; Borhany, M.; Guella, I.; Soldà, G.; Afrasiabi, A.; Menegatti, M.; Shamsi, T.; Pyvandi, F.; et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb. Haemost. 2016, 113, 567–576.
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and fibrin in hemostasis and thrombosis. Thromb. Vasc. Biol. 2017, 37, e13–e21.
- Beck, E.A.; Vogel, A.; Jackson, D.P. Functional evaluation of an inherited abnormal fibrinogen: Fibrinogen Baltimore. J. Clin. Investig. 1971, 50, 1874–1884.
- Kamijo, T.; Mukai, S.; Taira, C.; Higuchi, Y.; Okumura, N. γD318Y fibrinogen shows no fibrin polymerization due to defective “A-a” and “B-b” interactions, whereas that of γK321E fibrinogen is nearly normal. Thromb. Res. 2019, 182, 150–158.
- Borrell, M.; Gari, M.; Vallve, C.; Tirado, I.; Soria, J.M.; Sala, N.; Munoz, C.; Oliver, A.; Garcia, A.; Fontcuberta, J. Abnormal polymerization and normal binding of plasminogen and t-PA in three new dysfibrinogenemias: Barcelona III and IV (γArg 275→His) and Villajoyosa (γArg 275→Cys). Blood Coagul. Fibrinolysis 1995, 6, 198–206.
- Kotlin, R.; Reicheltova, Z.; Maly, M.; Suttnar, J.; Sobotkova, A.; Salaj, P.; Hirmerova, J.; Riedel, T.; Dyr, J.E. Two cases of congenital dysfibrinogenemia associated with thrombosis—Fibrinogen Prha III and Fibrinogen Plzen. Thromb. Haemost. 2009, 102, 479–486.
- Kaido, T.; Yoda, M.; Kamijo, T.; Taira, C.; Higuchi, Y.; Okumura, N. Comparison of molecular structure and fibrin polymerization between two Bβ—Chain N—terminal region fibrinogen variants, Bβp. G45C and Bβp.R74C. Int. J. Hematol. 2020, 112, 331–340.
- Fuss, C.; Palmaz, J.C.; Sprague, E.A. Fibringen: Structure, function, and surface interactions. J. Vasc. Interv. Radiol. 2001, 12, 677–682.
- Simurda, T.; Casini, A.; Stasko, J.; Hudecek, J.; Skornova, I.; Vilar, R.; Neerman-Arbez, M.; Kubisz, P. Perioperative mangement of a severe congenital hypofibrinogenemia with thrombotic phenotype. Thromb. Res. 2020, 188, 1–4.
- Besser, M.W.; MacDonald, S. Acquired hypofibrinogenemia: Current perspectives. J. Blood Med. 2016, 7, 217–225.
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192.
- Hanss, M.M.L.; Ffrench, P.O.; Morner, J.F.; Chabuet, M.; Biot, F.; De Mazancourt, M. The novel fibrinogen variants found in patients with pulmonary embolism and their families. J. Thromb. Haemost. 2003, 1, 1251–1257.
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76.
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital Fibrinogen Disorders: An Update. Semin. Thromb. Hemost. 2013, 39, 585–595.
- Asselta, R.; Spena, S.; Duga, S.; Tenchini, M.L. Molecular genetics of quantitative fibrinogen disorders. Cardiovasc Hematol. Agents Med. Chem. 2007, 5, 163–173.
- Hamano, A.; Mimuro, J.; Aoshima, M.; Itoh, T.; Kitamura, N.; Nishinarita, S.; Takano, K.; Ishiwata, A.; Kashiwakura, Y.; Niwa, K.; et al. Thrombophilic dysfibrinogen Tokyo V with the amino acid substitution of γAla327Thr: Formation of fragile but fibrinolysis—Resistant fibrin clots and its relevance to arterial thromboembolism. Blood 2004, 103, 3045–3050.
- Duval, C.; Ariens, R.S. Fibrinogen splice variation and cross-linking: Effects on fibrin structure/function and role of fibrinogen γ’as trhombomobulin II. Matrix Biol. 2017, 60, 8–15.
More
Information
Subjects:
Health Care Sciences & Services
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
02 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No