 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Stephan Bader | + 3457 word(s) | 3457 | 2022-02-09 04:40:48 | | | |
| 2 | Michael Stephan Bader | + 5 word(s) | 3462 | 2022-02-28 19:38:30 | | | | |
| 3 | Michael Stephan Bader | + 4 word(s) | 3461 | 2022-02-28 19:41:49 | | | | |
| 4 | Catherine Yang | -1 word(s) | 3460 | 2022-03-01 03:06:22 | | |
Video Upload Options
The discovery of the activating V617F mutation in Janus kinase 2 (JAK2) has been decisive for the understanding of myeloproliferative neoplasms (MPN). Activated JAK2 signaling by JAK2, CALR, and MPL mutations has become a focus for the development of targeted therapies for patients with MPN. JAK2 inhibitors now represent a standard of clinical care for certain forms of MPN and offer important benefits for MPN patients. However, several key aspects remain unsolved regarding the targeted therapy of MPN with JAK2 inhibitors, such as reducing the MPN clone and how to avoid or overcome a loss of response. The current knowledge on the structure and signaling of JAK2 as central elements of MPN pathogenesis and feature benefits and limitations of therapeutic JAK2 targeting in MPN were summarized.
1. Introduction
Janus kinases (JAKs) constitute a family of intracellular, non-receptor tyrosine kinases. They are associated with their corresponding cell surface receptors and transduce signals from cytokines, as well as some hormones, such as growth hormone and prolactin. Mediation of extracellular signals via JAKs activates intracellular messenger pathways, impacting hematopoiesis, metabolism, and immune responses. The discovery that most BCR-ABL1-negative myeloproliferative neoplasms (MPN) carry an activating JAK2 mutation has moved this tyrosine kinase into the center of interest and has led to refined diagnostic criteria for these disease entities, development of new prognostic models, and the introduction of JAK inhibitors into clinical practice.
2. JAK2 Structure and Signaling
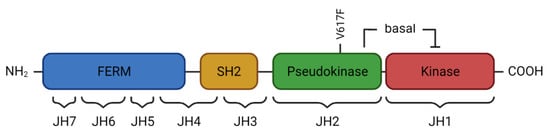
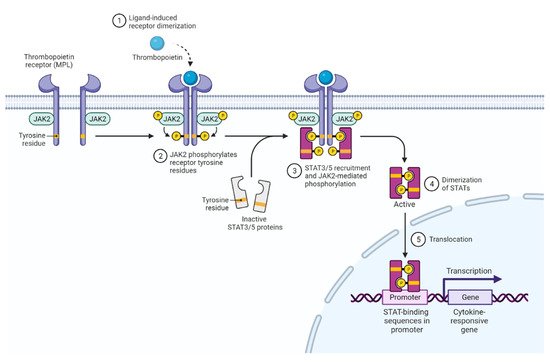
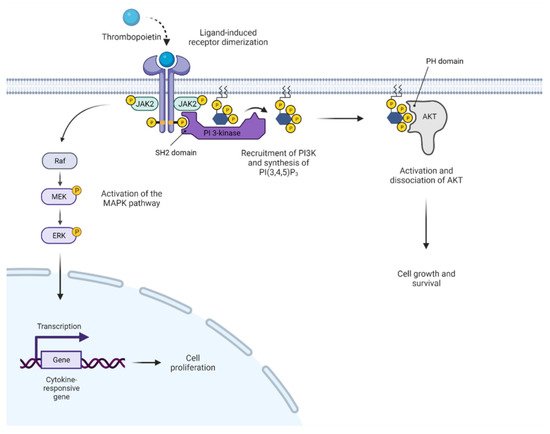
3. Activated JAK2 Signaling in MPN
4. Clinical Presentation of MPN
Philadelphia chromosome-negative myeloproliferative neoplasms (MPN) are clonal hematopoietic stem cell disorders and, based on activated JAK2 signaling, present with an excessive output of mature blood cells of one or more myeloid lineages [43]. When first described by William Dameshek in 1951, a hitherto unrevealed stimulus leading to proliferation of bone marrow cells was suggested to drive the MPN including PV, ET, and PMF [44]. While PV is characterized by erythrocytosis and potential thrombocytosis and neutrophilia, the hallmark of ET is isolated thrombocytosis. Primary myelofibrosis is characterized by enhanced, atypical megakaryopoiesis and cytoses in the pre-fibrotic phase and progressive bone marrow fibrosis and cytopenia in more advanced phases. Extramedullary hematopoiesis leading to hepatosplenomegaly is typical for all MPN and most pronounced in PMF, including the appearance of myeloid and erythroid precursors in the peripheral blood (leuko-erythroblastosis). Beyond the characteristic alterations of peripheral blood counts and bone marrow architecture, the JAK2, CALR, and MPL driver mutations have become biomarkers for the diagnosis of MPN and have been incorporated into the WHO classification of hematopoietic malignancies [44]. Specifically, the JAK2 V617F mutation is instrumental in differentiating polycythemia vera from secondary erythrocytosis. It is still incompletely understood how PV, ET, or MF phenotypes may arise upon presence of the same JAK2 V617F mutation. Several factors have been implicated, such as JAK2 V617F mutant allele burden, which is usually lower in ET and higher in PV and MF, as well as loss of heterozygosity by mitotic recombination at chromosome 9p, leading to homozygosity for JAK2 V617F. Furthermore, the acquisition order of the JAK2 V617F and additional non-driver mutations, such as in the TET2 gene, was shown to play a role in determining clinical MPN phenotypes [45][46][47].
The most frequent complications of MPN are thrombo-hemorrhagic events. MPN patients are at risk for both arterial and venous thromboses, affecting approximately 40% of patients with PV during the course of disease. Particularly, there is an increased susceptibility for thromboses in the hepato-lienal and portal venous systems, and splanchnic thrombosis are typically seen [48]. It has become clear that the prothrombotic condition in MPN relates to multiple factors including erythrocytosis, leukocytosis, activation of platelets, but also alterations in plasmatic coagulation and NET formation. In addition, inflammatory stimuli are known to induce endothelial dysfunction with pro-adhesive surface properties, which facilitates leukocyte and platelet binding and activation [49]. JAK2 V617F expression in endothelial cells appears to promote such functional changes and an overall inflammatory state of the vasculature [50]. Interestingly, JAK2 V617F is associated with a higher thrombotic risk than CALR mutations, and influences of the mutant cell fraction have also been shown [51]. Bleeding events are an additional concern and can relate to acquired von Willebrand syndrome due to preferential consumption of large von Willebrand factor (VWF) multimers in settings of excessive thrombocytosis with counts > 1000–1500 G/L.
While both PV and ET can progress to secondarymyelofibrosis referred to as Post-PV/Post-ET MF, the risk of transformation to secondary acutemyeloid leukemia (AML) is highest in PMF (10-year risk 10–20%), followed by PV (2–4%) and ET (approximately 1%) [52]. Outcomes are generally poor, reflecting the prevalent unfavorable cytogenetic andmolecular features, which render the leukemic clones less sensitive to standard chemotherapeutic agents. Leukemic transformation can arise from the JAK2 V617F mutant hematopoietic stem/progenitor compartment, but may also present as JAK2 V617F-negative AML. Leukemic blasts preferentially show mutations in one of the two isoforms of isocitrate dehydrogenase (IDH1, IDH2), the tumor suppressor gene TP53, or serine and arginine-rich splicing factor 2 (SRSF2) [53][54][55]. Risk of leukemic transformation is associated with the overall number of acquired mutations, and the vast majority of these are already detectable at MPN diagnosis [56].
5. Clinical Benefit of JAK Inhibitor Therapy in MPN
Constitutive activation of JAK2 signaling in JAK2, CALR, and MPL mutant settings as well as in triple-negative MPN has provided a rational basis to develop JAK2 inhibition as a therapeutic approach in MPN [57]. Current JAK2 inhibitors that are approved (ruxolitinib, fedratinib) or in clinical development (momelotinib, pacritinib, etc.) engage the ATP binding site of JAK2 in the active conformation, thereby interfering with JAK2 catalytic activity, and are referred to as type 1 inhibitors [58].
Ruxolitinib, which acts as a JAK1/2 inhibitor, represents a clinical standard of care for patients with intermediate or high-risk MF with symptomatic splenomegaly or constitutional symptoms. As demonstrated by the COMFORT trials, ruxolitinib is able to improve splenomegaly and symptom burden along with reduced cytoses and proinflammatory cytokine levels. A survival benefit was observed after one year of treatment [59][60]. Based on the RESPONSE study, ruxolitinib has also been approved for patients with PV resistance or intolerant to hydroxyurea [61] and is effectively reducing hematocrit, splenomegaly, and symptom burden. Ruxolitinib therapy also resulted in a lower frequency of thromboembolic events relating to reduced hematocrit and inflammation [62]. In ET, ruxolitinib has not shown additional clinical value so far [63].
Fedratinib, a second JAKinib with JAK2/FLT3 inhibitory activity, has recently been approved after clinical development was delayed by a clinical hold based on suspected cases of Wernicke’s encephalopathy. Analogous to ruxolitinib, fedratinib is able to reduce symptom burden and splenomegaly in JAK inhibitor-naïve patients as well as in patients who lose response or are intolerant to ruxolitinib, thus providing a second line JAK2 inhibitor treatment after ruxolitinib failure [64]. Monitoring of thiamine levels and/or supplementation is recommended and additional safety data from the phase 3b FREEDOM study are currently accumulating.
Several JAK2 inhibitors are in clinical development, which could prove beneficial in patients with anemia and/or thrombocytopenia. Pacritinib is a JAK2/FLT3 inhibitor with milder myelosuppressive properties and favorable results in patients with thrombocytopenia [65]. In addition, momelotinib represents a JAK1/2 inhibitor with additional impact on the activin A type 1 receptor (ACVR1), which favorably affects iron homeostasis and translates into reduced transfusion requirements [66][67]. Pacritinib and momelotinib are currently being evaluated in clinical trials and will hopefully soon extend our therapeutic options for cytopenic MPN patients.
Beyond MPN, the JAK1/2 inhibitor ruxolitinib has also shown activity in the therapy of glucocorticosteroid-refractory acute or chronic graft-versus-host disease [68][69]. Furthermore, immunosuppressive properties of JAK1/2 inhibitors are increasingly used to treatvarious autoimmune disorders, particularly rheumatoid arthritis.
6. Limitations of JAK Inhibitor Therapy in MPN
Current JAK2 inhibitor therapies in MPN show limited disease-modifying potential as an important limitation. Reductions in mutant allele burden are modest, and it has been demonstrated that clonal evolution is progressing during treatment with, e.g., ruxolitinib [70]. Continued therapy may lead to a certain decrease in clone size, but partial molecular remission, defined as a 50% reduction in JAK2 V617F allele burden in patients with at least 20% mutant allelic burden at baseline, is achieved in <10% of patients. Therefore, the development of more effective therapeutic approaches with disease-modifying potential is imperative.
Anemia, thrombocytopenia, and to a lesser extent immunosuppression represent frequent on-target toxicities of JAK inhibitor treatment and may require dose modifications [60][71]. While anemia is most pronounced at treatment initiation, it may stabilize over time.
JAK1 inhibitory activity of ruxolitinib has been related to reduced immune surveillance and the association with higher incidences of opportunistic infections and particularly reactivation of Herpes zoster, but also reactivation of tuberculosis, cryptococcal meningoencephalitis, Pneumocystis jirovecii pneumonia, hepatitis B, toxoplasmosis, or cytomegalovirus retinitis have been reported underscoring the importance of increased vigilance [72][73][74][75][76]. In addition, it has been speculated that MPN patients treated with ruxolitinib could be at higher risk for secondary cancers, and a trend for increased occurrence of non-melanoma skin cancer has been observed in long-term studies [77]. Furthermore, aggressive B-cell lymphomas have been reported, particularly in patients with MPN and preexisting B-cell clones, but the data are conflicting [78][79].
Loss of response to JAK inhibitor therapy can occur over time and is seen in approximately 50% of MF patients who initially responded to therapy after 5 years of treatment [80]. Second-site mutations inducing resistance by interference with drug binding have been described in MPN cell lines but have not been reported in MPN patients. Thus, resistance to JAK inhibitors appears to relate to adaptive processes, which is highlighted by the observation that patients can regain response upon re-exposure after a drug holiday [81]. While reactivation of JAK2 signaling via JAK family heterodimer formation has been described [82], additional mechanisms of acquired resistance to JAK2 inhibition have also been reported [83].
7. Potential Future Avenues for JAK2 Inhibitor Development and Alternative Therapeutic Approaches
8. JAK2 V617F in Clonal Hematopoiesis of Indeterminate Potential
References
- McNally, R.; Toms, A.V.; Eck, M.J. Crystal Structure of the FERM-SH2 Module of Human Jak2. PLoS ONE 2016, 11, e0156218.
- Raivola, J.; Haikarainen, T.; Abraham, B.G.; Silvennoinen, O. Janus Kinases in Leukemia. Cancers 2021, 13, 800.
- Wallweber, H.J.A.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural Basis of Recognition of Interferon-α Receptor by Tyrosine Kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448.
- Bandaranayake, R.M.; Ungureanu, D.; Shan, Y.; Shaw, D.E.; Silvennoinen, O.; Hubbard, S.R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19, 754–759.
- Saharinen, P.; Takaluoma, K.; Silvennoinen, O. Regulation of the Jak2 Tyrosine Kinase by Its Pseudokinase Domain. Mol. Cell. Biol. 2000, 20, 3387–3395.
- Argetsinger, L.S.; Kouadio, J.-L.K.; Steen, H.; Stensballe, A.; Jensen, O.N.; Carter-Su, C. Autophosphorylation of JAK2 on Tyrosines 221 and 570 Regulates Its Activity. Mol. Cell. Biol. 2004, 24, 4955–4967.
- Mazurkiewicz-Munoz, A.M.; Argetsinger, L.S.; Kouadio, J.-L.K.; Stensballe, A.; Jensen, O.N.; Cline, J.M.; Carter-Su, C. Phosphorylation of JAK2 at Serine 523: A Negative Regulator of JAK2 That Is Stimulated by GrowthHormone and Epidermal Growth Factor. Mol. Cell. Biol. 2006, 26, 4052–4062.
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The Regulation of JAKs in Cytokine Signaling and Its Breakdown in Disease. Cytokine 2019, 118, 48–63.
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The Molecular Regulation of Janus Kinase (JAK) Activation. Biochem. J. 2014, 462, 1–13.
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of Myeloproliferative Neoplasms. Exp. Hematol. 2015, 43, 599–608.
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384.
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007.
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-AKT-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907.
- Meyer, S.C.; Levine, R.L. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059.
- Varghese, L.N.; Ungureanu, D.; Liau, N.P.D.; Young, S.N.; Laktyushin, A.; Hammaren, H.; Lucet, I.S.; Nicola, N.A.; Silvennoinen, O.; Babon, J.J.; et al. Mechanistic Insights into Activation and SOCS3-Mediated Inhibition of Myeloproliferative Neoplasm-Associated JAK2 Mutants from Biochemical and Structural Analyses. Biochem. J. 2014, 458, 395–405.
- Xu, D.; Qu, C.K. Protein Tyrosine Phosphatases in the JAK/STAT Pathway. Front. Biosci. 2008, 13, 4925–4932.
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The Role of LNK/SH2B3 Genetic Alterations in Myeloproliferative Neoplasms and Other Hematological Disorders. Leukemia 2017, 31, 1661–1670.
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397.
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired Mutation of the Tyrosine Kinase JAK2 in Human Myeloproliferative Disorders. Lancet 2005, 365, 1054–1061.
- Ugo, V.; James, C.; Vainchenker, W. A Unique Clonal JAK2 Mutation Leading to Constitutive Signalling Causes Polycythaemia Vera. Medicine/Sciences 2005, 21, 669–670.
- Shan, Y.; Gnanasambandan, K.; Ungureanu, D.; Kim, E.T.; Hammarén, H.; Yamashita, K.; Silvennoinen, O.; Shaw, D.E.; Hubbard, S.R. Molecular Basis for Pseudokinase-Dependent Autoinhibition of JAK2 Tyrosine Kinase. Nat. Struct. Mol. Biol. 2014, 21, 579–584.
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236.
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468.
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and Clinical Features of the Myeloproliferative Neoplasm Associated with JAK2 Exon 12 Mutations. Blood 2011, 117, 2813–2816.
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, 1140–1151.
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 Mutations in Myeloproliferative and Other Myeloid Disorders: A Study of 1182 Patients. Blood 2006, 108, 3472–3476.
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390.
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405.
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381.
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin Receptor Activation by Myeloproliferative Neoplasm Associated Calreticulin Mutants. Blood 2016, 127, 1325–1335.
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR Mutation Status Defines Subtypes of Essential Thrombocythemia with Substantially Different Clinical Course and Outcomes. Blood 2014, 123, 1544–1551.
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated Genomic Analysis Illustrates the Central Role of JAK-STAT Pathway Activation in Myeloproliferative Neoplasm Pathogenesis. Blood 2014, 123, e123–e133.
- Rinaldi, C.R.; Rinaldi, P.; Alagia, A.; Gemei, M.; Esposito, N.; Formiggini, F.; Martinelli, V.; Senyuk, V.; Nucifora, G.; Pane, F. Preferential Nuclear Accumulation of JAK2V617F in CD34+ but Not in Granulocytic, Megakaryocytic, or Erythroid Cells of Patients with Philadelphia-Negative Myeloproliferative Neoplasia. Blood 2010, 116, 6023–6026.
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 Phosphorylates Histone H3Y41 and Excludes HP1α from Chromatin. Nature 2009, 461, 819–822.
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294.
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2V617F-Mutant MPN. Cancer Discov. 2020, 10, 1742–1757.
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Review Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561.
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα Facilitates Clonal Expansion of JAK2V617F Positive Cells in Myeloproliferative Neoplasms. Blood 2011, 118, 6392–6398.
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A Role for Reactive Oxygen Species in JAK2 V617F Myeloproliferative Neoplasm Progression. Leukemia 2013, 27, 2187–2195.
- Hasselbalch, H.C. The Role of Cytokines in the Initiation and Progression of Myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145.
- Găman, M.A.; Cozma, M.A.; Dobrică, E.C.; Crețoiu, S.M.; Găman, A.M.; Diaconu, C.C. Liquid Biopsy and Potential Liquid Biopsy-Based Biomarkers in Philadelphia-Negative Classical Myeloproliferative Neoplasms: A Systematic Review. Life 2021, 11, 677.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405.
- DAMESHEK, W. Some Speculations on the Myeloproliferative Syndromes. Blood 1951, 6, 372–375.
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of Myeloproliferative Neoplasms. Exp. Hematol. 2015, 43, 599–608.
- Bandaranayake, R.M.; Ungureanu, D.; Shan, Y.; Shaw, D.E.; Silvennoinen, O.; Hubbard, S.R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19, 754–759.
- Saharinen, P.; Takaluoma, K.; Silvennoinen, O. Regulation of the Jak2 Tyrosine Kinase by Its Pseudokinase Domain. Mol. Cell. Biol. 2000, 20, 3387–3395.
- Falanga, A.; Marchetti, M. Thrombosis in Myeloproliferative Neoplasms. Semin. Thromb. Hemost. 2014, 40, 348–358.
- Shao, Y.; Cheng, Z.; Li, X.; Chernaya, V.; Wang, H.; Yang, X.F. Immunosuppressive/Anti-Inflammatory Cytokines Directly and Indirectly Inhibit Endothelial Dysfunction-A Novel Mechanism for Maintaining Vascular Function. J. Hematol. Oncol. 2014, 7, 80.
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.A.; Denis, C.; Villeval, J.L.; et al. Vascular Endothelial Cell Expression of JAK2 V617F Is Sufficient to Promote a Pro-Thrombotic State Due to Increased P-Selectin Expression. Haematologica 2019, 104, 70–81.
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Lo Coco, F.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of Patients with Poorer Survival in Primary Myelofibrosis Based on the Burden of JAK2V617F Mutated Allele. Blood 2009, 114, 1477–1483.
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-Term Survival and Blast Transformation in Molecularly Annotated Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis. Blood 2014, 124, 2507–2513.
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 Mutation Studies in 1473 Patients with Chronic-, Fibrotic- or Blast-Phase Essential Thrombocythemia, Polycythemia Vera or Myelofibrosis. Leukemia 2010, 24, 1302–1309.
- Zhang, S.J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic Analysis of Patients with Leukemic Transformation of Myeloproliferative Neoplasms Shows Recurrent SRSF2 Mutations That Are Associated with Adverse Outcome. Blood 2012, 119, 4480–4485.
- Rampal, R.; Ahn, J.; Abdel-Wahaba, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and Functional Analysis of Leukemic Transformation of Myeloproliferative Neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410.
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal Evolution and Clinical Correlates of Somatic Mutations in Myeloproliferative Neoplasms. Blood 2014, 123, 2220–2228.
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin Receptor Activation by Myeloproliferative Neoplasm Associated Calreticulin Mutants. Blood 2016, 127, 1325–1335.
- Brkic, S.; Meyer, S.C. Challenges and Perspectives for Therapeutic Targeting of Myeloproliferative Neoplasms. Hemasphere 2021, 5, e516.
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807.
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-Term Findings from COMFORT-II, a Phase 3 Study of Ruxolitinib vs Best Available Therapy for Myelofibrosis. Leukemia 2016, 30, 1701–1707.
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N. Engl. J. Med. 2015, 372, 426–435.
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. CYTO-PV Collaborative Group. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. N. Engl. J. Med. 2013, 368, 22–33.
- Harrison, C.N.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib vs. Best Available Therapy for et Intolerant or Resistant to Hydroxycarbamide. Blood 2017, 130, 1889–1897.
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients with Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651.
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients with Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659.
- Oh, S.T.; Talpaz, M.; Gerds, A.T.; Gupta, V.; Verstovsek, S.; Mesa, R.; Miller, C.B.; Rivera, C.E.; Fleischman, A.G.; Goel, S.; et al. ACVR1/JAK1/JAK2 Inhibitor Momelotinib Reverses Transfusion Dependency and Suppresses Hepcidin in Myelofibrosis Phase 2 Trial. Blood Adv. 2020, 4, 4282–4291.
- Gupta, V.; Mesa, R.A.; Deininger, M.W.N.; Rivera, C.E.; Sirhan, S.; Brachmann, C.B.; Collins, H.; Kawashima, J.; Xin, Y.; Verstovsek, S. A Phase 1/2, Open-Label Study Evaluating Twice-Daily Administration of Momelotinib in Myelofibrosis. Haematologica 2017, 102, 94–102.
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810.
- Zeiser, R.; Polverelli, N.; Ram, R.; Hashmi, S.K.; Chakraverty, R.; Middeke, J.M.; Musso, M.; Giebel, S.; Uzay, A.; Langmuir, P.; et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease. N. Engl. J. Med. 2021, 385, 228–238.
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal Evolution and Outcomes in Myelofibrosis after Ruxolitinib Discontinuation. Blood 2017, 130, 1125–1131.
- Kiladjian, J.J.; Zachee, P.; Hino, M.; Pane, F.; Masszi, T.; Harrison, C.N.; Mesa, R.; Miller, C.B.; Passamonti, F.; Durrant, S.; et al. Long-Term Efficacy and Safety of Ruxolitinib versus Best Available Therapy in Polycythaemia Vera (RESPONSE): 5-Year Follow up of a Phase 3 Study. Lancet Haematol. 2020, 7, e226–e237.
- Luo, Q.; Xiao, Z.; Peng, L. Effects of Ruxolitinib on Infection in Patients with Myeloproliferative Neoplasm: A Meta-Analysis. Hematology 2021, 26, 663–669.
- Sadjadian, P.; Wille, K.; Griesshammer, M.; Sadjadian, P.; Wille, K.; Griesshammer, M. Ruxolitinib-Associated Infections in Polycythemia Vera: Review of the Literature, Clinical Significance, and Recommendations. Cancers 2020, 12, 3132.
- Khalid, F.; Damlaj, M.; AlZahrani, M.; Abuelgasim, K.A.; Gmati, G.E. Reactivation of Tuberculosis Following Ruxolitinib Therapy for Primary Myelofibrosis: Case Series and Literature Review. Hematol. Oncol. Stem Cell Ther. 2021, 14, 252–256.
- Duan, M.-H.; Cao, X.-X.; Chang, L.; Zhou, D.-B. Risk of Hepatitis B Virus Reactivation Following Ruxolitinib Treatment in Patients with Myeloproliferative Neoplasms. Hematology 2021, 26, 460–464.
- Devos, T.; Selleslag, D.; Granacher, N.; Havelange, V.; Benghiat, F.S. Updated Recommendations on the Use of Ruxolitinib for the Treatment of Myelofibrosis. Hematology 2022, 27, 23–31.
- Barraco, F.; Greil, R.; Herbrecht, R.; Schmidt, B.; Reiter, A.; Willenbacher, W.; Raymakers, R.; Liersch, R.; Wroclawska, M.; Pack, R.; et al. Real-World Non-Interventional Long-Term Post-Authorisation Safety Study of Ruxolitinib in Myelofibrosis. Br. J. Haematol. 2020, 191, 764–774.
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-Cell Lymphomas in Patients with Myelofibrosis Receiving JAK1/2 Inhibitor Therapy. Blood 2018, 132, 694–706.
- Sekhri, R.; Sadjadian, P.; Becker, T.; Kolatzki, V.; Huenerbein, K.; Meixner, R.; Marchi, H.; Wallmann, R.; Fuchs, C.; Griesshammer, M.; et al. Ruxolitinib-Treated Polycythemia Vera Patients and Their Risk of Secondary Malignancies. Ann. Hematol. 2021, 100, 2707–2716.
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-Term Treatment with Ruxolitinib for Patients with Myelofibrosis: 5-Year Update from the Randomized, Double-Blind, Placebo-Controlled, Phase 3 COMFORT-I Trial. J. Hematol. Oncol. 2017, 10, 55.
- Gisslinger, H.; Schalling, M.; Gisslinger, B.; Skrabs, C.; Müllauer, L.; Kralovics, R. Restoration of Response to Ruxolitinib upon Brief Withdrawal in Two Patients with Myelofibrosis. Am. J. Hematol. 2014, 89, 344–346.
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT Activation as a Mechanism of Persistence to JAK2 Inhibitor Therapy. Nature 2012, 489, 155–159.
- Meyer, S.C. Mechanisms of Resistance to JAK2 Inhibitors in Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2017, 31, 627–642.
- Meyer, S.C.; Keller, M.D.; Chiu, S.; Koppikar, P.; Guryanova, O.A.; Rapaport, F.; Xu, K.; Manova, K.; Pankov, D.; O’Reilly, R.J.; et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015, 28, 15–28.
- Wu, S.; Li, L.S.; Kopp, N.; Montero, J.; Chapuy, B.; Yoda, A.; Christie, A.L.; Liu, H.; Christodoulou, A.; van Bodegom, D.; et al. Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 29–41.
- Hammarén, H.M.; Ungureanu, D.; Grisouard, J.; Skoda, R.C.; Hubbard, S.R.; Silvennoinen, O. ATP Binding to the Pseudokinase Domain of JAK2 Is Critical for Pathogenic Activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647.
- Harrison, C.; Garcia, J.S.; Mesa, R.; Somervaille, T.; Ritchie, E.K.; Komrokji, R.S.; Pemmaraju, N.; Jamieson, C.; Papadantonakis, N.; Foran, J.M.; et al. MPN-038: Navitoclax in Combination with Ruxolitinib in Patients with Primary or Secondary Myelofibrosis: A Phase 2 Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, S325.
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.S.; Bracken, L.S.; et al. Combined Targeting of JAK2 and Bcl-2/Bcl-XL to Cure Mutant JAK2-Driven Malignancies and Overcome Acquired Resistance to JAK2 Inhibitors. Cell Rep. 2013, 5, 1047–1059.
- Yacoub, A.; Borate, U.; Rampal, R.; Ali, H.; Wang, E.; Gerds, A.; Hobbs, G.; Kremyanskaya, M.; Winton, E.; O’Connell, C.; et al. MPN-127: Add-On Parsaclisib (a PI3K5 Inhibitor) in Patients with Myelofibrosis and Suboptimal Response to Ruxolitinib: Interim Analysis from a Phase 2 Study. Clin. Lymphoma Myeloma Leuk. 2021, 21, S354–S355.
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting Compensatory MEK/ERK Activation Increases JAK Inhibitor Efficacy in Myeloproliferative Neoplasms. J. Clin. Investig. 2019, 129, 1596–1611.
- Brkic, S.; Stivala, S.; Santopolo, A.; Szybinski, J.; Jungius, S.; Passweg, J.R.; Tsakiris, D.; Dirnhofer, S.; Hutter, G.; Leonards, K.; et al. Dual Targeting of JAK2 and ERK Interferes with the Myeloproliferative Neoplasm Clone and Enhances Therapeutic Efficacy. Leukemia 2021, 35, 2875–2884.
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing Factor YBX1 Mediates Persistence of JAK2-Mutated Neoplasms. Nature 2020, 588, 157–163.
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121.
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-Related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 2014, 20, 1472–1478.
- Patel, R.K.; Lea, N.C.; Heneghan, M.A.; Westwood, N.B.; Milojkovic, D.; Thanigaikumar, M.; Yallop, D.; Arya, R.; Pagliuca, A.; Gäken, J.; et al. Prevalence of the Activating JAK2 Tyrosine Kinase Mutation V617F in the Budd-Chiari Syndrome. Gastroenterology 2006, 130, 2031–2038.


