 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David N. Brindley | + 3690 word(s) | 3690 | 2020-09-03 10:18:57 | | | |
| 2 | Camila Xu | Meta information modification | 3690 | 2020-09-11 04:15:40 | | |
Video Upload Options
Lipid phosphate phosphatases (LPPs) consist of three enzymes (LPP1–3), which have been classified as phospholipid phosphatases (PLPP). The LPPs dephosphorylate a wide spectrum of bioactive lipid phosphates, among which lysophosphatidate (LPA) and sphingosine 1-phosphate (S1P) are two important extracellular signaling molecules. The LPPs are integral membrane proteins, which are partly localized on plasma membranes. These activities participate in regulating the concentrations of extracellular LPA and S1P and thus signaling through their families of G protein coupled receptors. The expression of the LPPs on intracellular membranes, including endoplasmic reticulum and Golgi net work, are thought to attenuate signaling downstream of the activation of LPA and protease activates receptors. The LPPs differentially regulate signal transduction in cancer cells. LPP1 and LPP3 have decreased expressions in several caners and this is associated with increased tumor growth and metastasis. Conversely, LPP2 activity is increased in these cancers and this accelerates progression through the cell cycle. Increasing the relative activities of LPP1 and LPP3 and decreasing LPP2 activity, therefore, provides a novel approach to treating some cancers.
1. Introduction
Mammalian LPP1–3 are encoded by three separate genes, PLPP1, PLPP2, and PLPP3, and they hydrolyze a wide spectrum of lipid phosphates including phosphatidate (PA), lysophosphatidate (LPA), sphingosine 1-phosphate (S1P), ceramide 1-phosphate (C1P), and diacylglycerol pyrophosphate (DGPP) in a Mg2+-independent and N-ethylmaleimide (NEM)-insensitive manner [1,2]. PLPP4 and PLPP5 are the former diacylglycerol pyrophosphate phosphatase-like 2 (DPPL2) and 1 (DPPL1), respectively. PLPP4–5 prefer DGPP as a substrate and also hydrolyze PA and LPA [3]. The activities of PLPP4–5 are also Mg2+-independent, but they can be inhibited by NEM [3]. PLPP6 is formerly known as polyisoprenyl diphosphate phosphatase 1 (PDP1) or candidate sphingomyelin synthase type 2β (CSS2β), which hydrolyzes presqualene diphosphate (PSDP), farnesyl diphosphate (FDP), S1P, LPA, and PA, but it has a preference for PSDP [4,5]. LPPs (PLPP1–3) and PLPP4–6 share highly conserved catalytic domains but show different substrate preferences. LPPs are responsible for the breakdown of extracellular LPA and S1P, which are two important signal molecules and therefore participate in many physiological and pathological processes such as vascular development [6], cell cycle regulation [7], cardiovascular disease [8], and cancer [9]. So far, there are very few reports about PLPP4–6, and their physiological functions are not clear. PLPP7, formerly known as NET39 or CSS2α, is catalytically inactive as a phosphatase due to the loss of critical amino acids in the catalytic domains [10,11].
The process of identifying LPPs dates back to the 1950s when a phosphatidate phosphatase (PAP) activity that dephosphorylates PA to form diacylglycerol (DAG) was discovered in mammalian tissue [12,13]. The PAP activity was intensively investigated as a critical regulator of lipid metabolism because the transformation from PA to DAG represents an intermediate reaction in the Kennedy pathway [14]. Early studies found that the cytosolic and membrane-bound PAPs exhibit quite different enzymological characteristics. For instance, the activity of the cytosolic PAP (PAP-1) that translocates onto membranes of the endoplasmic reticulum (ER) depends on the presence of Mg2+ and is sensitive to NEM [15,16,17,18]. Its activity is required for the synthesis of triacylglycerol, phosphatidylcholine, and phosphatidylethanolamine [19,20]. It was not until 2006 that PAP-1 was identified in yeast and was found to be the orthologue of a family of three mammalian proteins called lipins [21]. Then, all three of the mammalian lipins were shown to have PAP activity, which is involved in glycerolipid synthesis [22].
A Mg2+-independent phosphatidate phosphatase activity (PAP-2) was also described, and this activity was found mainly in the plasma membrane fraction [23]. This activity in mammals was not inhibited by NEM, which further distinguished it from PAP-1 activity. This new class of PAP activities was characterized in liver [23,24,25,26]. Unlike PAP-1, which is specific for PA, PAP-2 degrades a wide spectrum of phospholipids including PA, LPA, S1P, C1P, and lipid pyrophosphates in vitro [1]. This observation led to the more accurate naming of the PAP-2 activity as a lipid phosphate phosphatases [27]. The identification of PAP-2 at a molecular level was achieved by the revelation of cDNA sequences of three PAP-2 isoforms (PAP-2a, PAP-2b, and PAP-2c) in human beings and other animals [28,29,30,31]. These isoforms share amino acid sequence homology, and LPP orthologs were also identified in fruit flies and yeast [32,33,34].
mRNA of LPP1–3 are universally expressed in different tissues of human beings including adrenal, appendix, bone marrow, brain, colon, duodenum, endometrium, esophagus, fat, gall bladder, heart, kidney, liver, lung, lymph node, ovary, pancreas, placenta, prostate, salivary gland, skin, small intestine, spleen, stomach, testis, thyroid, and urinary bladder [35]. Protein expression data from The Human Protein Atlas (http://www.proteinatlas.org) indicate that LPP1–3 are expressed in most tissues, among which LPP1 is highly expressed in the prostate and kidney. LPP2 is expressed at a higher level in the gastrointestinal tract, salivary gland, gallbladder, pancreas, kidney, urinary bladder, and brain than in other tissue, while LPP3 is high in lung, salivary gland, oral mucosa, duodenum, smooth muscle, and skin [36].
2. Structure and Membrane Topology of LPP
The mammalian LPPs are localized on the plasma membrane and intracellular network of ER and Golgi [7,37]. It has been reported that LPP1 and LPP3 are present in lipid rafts or caveolae [38,39]. There is also evidence that LPP1 can be directed to the apical surface membrane by a FDKTRL motif on the N-terminus, whereas LPP3 is accumulated at the basolateral membrane [40]. The crystal structure of the LPPs has not yet been solved. A putative topology for the LPPs was determined based on the data obtained from hydrophobicity plots and transmembrane disposition analysis of the rat Dri42 protein [41], which later proved to be rat LPP3 [28]. It has six membrane-spanning regions connected by five extramembrane loops (I–V). Both C- and N-terminal extensions and loop II and IV are located in the cytosol. Loops I, III, and V are on the extracellular side of the membrane. (Figure 1). Three conserved domains (C1, C2, and C3) that form the catalytic site are located on loops III and V outside the cells. Residues that are indispensable for the phosphatase activity in C1–C3 (Figure 2) were identified by amino acid substitution analysis [42]. LPPs inside the cells are localized in the ER [37,41] and Golgi [28]. There is an N-linked glycosylation site between C1 and C2 (Figure 1) [42], indicating that the catalytic site are on the luminal side of ER and Golgi where LPPs are glycosylated [42]. This topology enables LPPs to hydrolyze substrates outside of the cells and in the lumen of ER and Golgi [9,43].
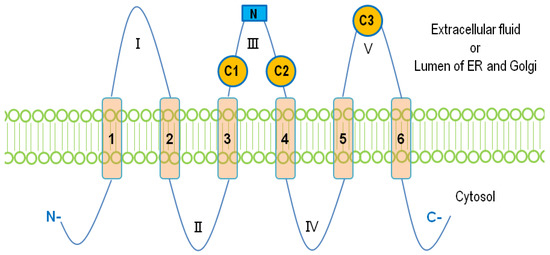
Figure 1. The membrane topology of lipid phosphate phosphatases (LPPs). Six membrane-spanning regions (1–6) are connected with five extramembrane loops (I–V). Three conserved catalytic domains, C1, C2, and C3, are located on loops III and V. The N-linked glycosylation site on the loop III is shown as a blue square.

Figure 2. Amino acid sequences of the conserved catalytic domains, C1, C2, and C3, in human LPPs and other proteins with structure similarity. Residues critical for the catalytic activity are shown in red.
The catalytic mechanism of LPPs has been postulated and proposed through a combination of computational modeling and the crystal structure of chloroperoxidase, which is a related enzyme that also possesses the C1–3 domains [44,45]. The conserved histidine on C3 serves as the nucleophile acting on the phosphate group to form a phospho-histidine intermediate. The C2 histidine is involved in breaking the phosphate bond. The conserved lysine and arginine on C1 as well as the arginine on C3 help coordinate the substrate in the active site [43,44,45]. Similar domains are also found in PLPP4–7. Unlike PLPP1–3, PLPP6 only has four transmembrane helices, and C1–3 of PLPP6 are located at the cytosolic side of the membrane. This allows PLPP6 to hydrolyze polyisoprenoid diphosphates in the cytosol [46]. Sphingosine phosphate phosphatases (SPPs), sphingomyelin synthases (SMSs), phospholipid phosphatase-related proteins (PLPPRs) [43,47], glucose 6-phosphatase (G6P), and E. coli phosphatidylglycerol-phosphate phosphatase B (PGPB), an orthologue of human G6P [33], also have the conserved catalytic domains. It is notable that the putative structure of PGPB was established through its crystal structure, which was determined later [47]. The structure of LPPs is thought to be modeled accurately from that proposed for PGPB.
3. Ecto-Activity of LPPs
A major part of circulating LPA is generated from lysophosphatidylcholine (LPC) through the lysophospholipase D activity of autotaxin (ATX) [48,49]. LPC is abundant in circulation with a concentration (>200 μM in human beings) [50], which is much higher than the Km of ATX for LPC (approximately 100 μM) [51]. As a secretary enzyme, ATX can readily access the LPC pool to generate LPA.
S1P is a sphingolipid analogue of LPA. The precursor for S1P synthesis is sphingosine, which is formed through the hydrolysis of ceramide by ceramidases. Sphingosine is phosphorylated by sphingosine kinase-1 and -2 (SPHK1 and 2) inside cells to generate S1P. SPHK1 is cytosolic and it interacts with the plasma membrane, whereas SPHK2 is present in the mitochondria [52] and nuclei [53]. S1P can be exported out the cells by the membrane transporters including ATP-binding cassette (ABC) transporters (ABCC1, ABCG2, and ABCA1) [54,55,56,57], spinster homolog-2 (SPNS2) [58], and major facilitator superfamily transporter 2b (Mfsd2b) [59]. This facilitates the “inside-out signaling” of S1P [54].
Both LPA and S1P outside the cells induce a plethora of cellular responses such as proliferation, migration, angiogenesis, and inflammation [51,60,61,62] through receptors on the cell surface. To date, six LPA receptors (LPAR1–6) and five S1P receptors (S1PR1–5) have been identified and all of them are G protein-coupled receptors (GPCRs). Plasma membrane-localized LPPs dephosphorylate extracellular LPA and S1P and thereby attenuate LPA/S1P signaling.
The ecto-phosphatase activity was established in rat2 fibroblasts where the overexpression of LPP1 increased the dephosphorylation of extracellular LPA, PA, and C1P. This action attenuated LPA-induced MAPK (mitogen-activated protein kinase) activation and inhibited cell migration [37,63,64]. Similarly, LPP1 and LPP2 inhibited the activation of MAPK that was stimulated by LPA or S1P in HEK293 cells [65]. The dephosphorylation of LPA generates monoacylglycerol (MAG), which can be transported into the cells and re-phosphorylated to form intracellular LPA [66]. This intracellular LPA can activate LPA1 receptors on the nuclear membrane and stimulate the expression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) [67]. Intracellular LPA has also been reported to initiate signaling through peroxisome proliferator-activated receptor γ (PPARγ) [68].
The ecto-activity of LPPs in vivo is more complex. Exogenous LPA injected into the circulation is turned over rapidly with the half-life of approximately 1 min [69]. LPP1 knockout mice showed increased levels and a decreased turnover rate of circulating LPA [70]. A similar phenotype was observed in LPP1 hypomorph mice, which have a low expression of LPP1 in most organs except the brain [71]. Interestingly, mice that transgenically overexpressed LPP1 did not show a decrease in the circulating LPA concentrations [72], suggesting that other factors may affect the ecto-activity of LPPs in vivo. For instance, the activity of LPPs is inhibited strongly by Ca2+, which is present in the extracellular environment at approximately 2 mM [37]. In addition, the physiological concentration of LPA in the plasma (0.1–1 µM) is much lower than the Km of LPP1 for LPA (approximately 36 μM) [37]. This indicates that the ecto-activity of LPPs is more important when the LPA levels are increased. In cancers, extracellular LPA levels are elevated as high as 10 μM [73,74,75]. We do not know if LPA concentrations in the vicinity of the LPPs are modified by other factors such as the levels of expression of the LPA receptors.
S1P concentrations in the plasma range from 100 nM to 1 μM [54]. Exogenous S1P injected into the circulation is cleared from the blood in 15–30 min [76]. S1P is dephosphorylated by SPPs and LPPs, or irreversibly cleaved by S1P lyase (SPL). SPPs and SPL are localized on the ER [77,78]. Therefore, the plasma membrane-localized LPPs have an essential role in regulating the amount of extracellular S1P. The ecto-activity of LPP1 and LPP3 against S1P has been demonstrated in cells [69,79] and animals [80,81]. LPP3 and LPP1a, a splice variant of LPP1, seem to be more efficient at hydrolyzing S1P than LPP1 and LPP2. Phospho-FTY720, an analogue of S1P, can be converted to FTY720 by LPP3 and LPP1a [82], but not by LPP1 or LPP2 [83]. Similarly, expressing exogenous LPP1, LPP2, or LPP3 in HEK293 cells enhances the ecto-activity against LPA, but only LPP3 significantly increases the degradation of extracellular S1P [69]. Sphingosine formed by the dephosphorylation of S1P can be transported into the cells and re-phosphorylated into S1P [79]. Therefore, this process represents a mechanism for the entry of S1P into cells.
4. Intracellular Activities of the LPPs
Not all of the effects of LPPs can be attributed to their ecto-activities. LPP1 is able to suppress wls-31-induced cell migration and Ca2+ mobilization [64,84]. Wls-31 is an isosteric phosphonate analog of LPA that activates LPAR1/2, but cannot be hydrolyzed by LPPs. LPP1 and LPP2 also inhibit MAPK activation induced by thrombin, which activates protease-activated receptors (PARs) [65]. Similarly, Ca2+ mobilization induced by a PAR1 peptide in MDA-MB-231 cells is inhibited by increased LPP1 expression [84]. Furthermore, LPP1 decreases the platelet derived growth factor (PDGF)-induced migration of embryonic fibroblasts through inhibiting the PDGF/PKC (protein kinase C) /MAPK pathway [63]. These effects of the LPPs are independent of their ecto-activities because these agonists cannot be degraded by LPPs. However, the effect requires LPP activity and therefore probably depends on the degradation of an intracellular lipid phosphate that is formed downstream of the activation of LPA, PAR, or PDGF receptors.
LPPs are also present on the ER and Golgi network with the catalytic domains, which should face the luminal side. As such, LPPs probably have specific access to substrates depending on the subcellular compartment. One of these possible substrates inside the cells is PA, which activates Sos (son of sevenless), Raf (rapidly accelerated fibrosarcoma), MAPK, mTOR (mammalian target of rapamycin), AKT (Ak strain transforming), and SPHK1 [85,86,87]. The dephosphorylation of PA generates DAG, which activates the classical and novel PKCs and Ras (rat sarcoma) guanyl nucleotide-releasing protein [88]. Increasing LPP1, LPP2, or LPP3 does decrease intracellular PA/DAG ratios [38,89]. LPP3 depletion decreases the levels of de novo synthesized DAG and the Golgi-associated DAG [90]. LPP2 decreases intracellular PA, which promotes the apoptosis of HEK293 cells in serum-deprived media [91]. However, LPP3 or LPP1 did not change intracellular DAG significantly in other studies [65,72,92].
Since the catalytic domains of LPPs are on the luminal side of ER and Golgi or the outer surface of the plasma membrane, the LPPs should not be able to dephosphorylate PA, which is formed at the cytosolic side of the membranes, unless the PA can be transported across the membranes to the catalytic sites of LPPs. However, this has yet to be shown. It should be noted that increasing LPP1 activity directly inhibits phospholipase D (PLD) activation [64], which forms a large proportion of intracellular PA. This can provide an alternative explanation for the decreased accumulation of PA. It is likely that the lipins, which are cytosolic phosphatidate phosphatases that translocate to membranes, are responsible for the degradation of most of the PA on the cytosolic surface of membranes [93].
LPPs probably also degrade intracellular C1P and S1P, both of which are involved in inflammation. C1P activates phospholipase A2 (PLA2) to produce arachidonate, which is converted to inflammatory eicosanoids (prostaglandins and thromboxanes) by COX-1/2 [94]. S1P helps to coordinate the metabolism of arachidonate by COX-2 to ensure the maximum production of prostaglandin E2 (PGE2) [94]. S1P also interacts with specific intracellular target proteins such as histone deacetylase 1/2, prohibitin 2, PPARγ, and tumor necrosis factor (TNF) receptor associated factor 2, to induce cell responses [95]. The overexpression of LPP3, but not LPP2, decreases intracellular S1P in HEK293 cells [91]. The degradation of intracellular S1P can be performed by other enzymes such as S1P phosphatases and S1P lyase, which are major regulators of intracellular S1P concentrations.
5. Upregulation of LPA Signaling in Cancers
Functioning as a platelet activator, a chemoattractant, and a growth factor, LPA plays a critical role in wound healing [96]. At sites of tissue damage, LPA stimulates the proliferation of fibroblasts and endothelial cells [97], and it promotes collagen deposition [98] and angiogenesis [99,100]. Circulating LPA concentrations are normally between 0.1 and 1 μM [51], and this is regulated mainly by the balance of ATX activity versus that of the LPPs.
LPA signaling is magnified and hijacked by cancers (wounds that do not heal) [101]. Elevated ATX levels have been observed in the blood and malignant tissues from patients with thyroid [102], lung [103], breast [104], liver [105], pancreatic [106,107], kidney [108], bladder [108], and prostate cancer [109]. As a consequence, LPA levels increase in those cancers [107,110,111,112,113], which has been considered an indicator of poor prognosis [110,113]. Significantly, LPA concentrations have been reported to reach as high as 10 μM in the ascites fluid of ovarian cancer patients [73,74,75]. Cancer cells express high levels of LPAR1–3 [61], which are GPCRs. LPAR1–3 couple to G proteins: Gi/o, Gq/11, and G12/13 [61], and activate PI3K (phosphoinositide 3-kinase) /AKT [114,115], PLC (phospholipase C) [116], and Rho pathways [116]. LPAR1–3 are elevated in brain [117,118], pancreatic [119,120,121], colon [122,123], and breast cancer [124], which is associated with enhanced tumor growth and metastasis.
Introducing exogenous LPAR1 converts non-transformed MCF-10A cells into an invasive phenotype [125]. LPAR1 and/or LPAR3 activate Wnt/β-catenin and PI3K/AKT/mTOR pathways to induce the epithelial-to-mesenchymal transition (EMT) [126,127], which is an essential step during cancer cell stemness [128]. Cancer stem cell (CSC)-related genes such as ALDH1A1, OCT4, and SOX2 are upregulated by activating LPAR1 [129]. Blocking ATX or LPAR2 suppresses the growth of breast cancer stem cells [62,130] in which LPP3 expression is downregulated [131]. Transgenic mice overexpressing ATX or any of LPAR1–3 by MMTV-LTR (mouse mammary tumor virus long terminal repeat) promoter in mammary epithelial cells show an increased development of spontaneous breast tumors and subsequent metastases [132]. LPAR4–6 are closely related to purinergic receptors [61]. LPAR4 (P2Y9/GPR23) and LPAR5 (GPR92) in cancer cells demonstrate inhibitory effects on proliferation and migration/invasion [133,134,135,136], which is in contrast to the effects of LPAR1–3. It is notable that LPAR5 suppresses the function of infiltrated CD8+ cytotoxic T cells as a mediator of immune suppression in the tumor microenvironment (TME) [137]. The effects of LPAR6 (P2Y5) in cancers are uncertain [138,139] and require further investigation.
LPA induces lymphocyte homing [140] and the transformation of monocytes to macrophages [141], which provokes inflammation [102,142]. LPA is closely related to the inflammatory milieu in conditions such as pulmonary fibrosis, rheumatoid arthritis, atherosclerosis, and inflammatory bowel disease [143]. The TME is also characterized by chronic inflammation, which is one of the hallmarks of cancers [144]. Increasing evidence reveals that there is crosstalk between LPA signaling and cancer-related inflammation. TNFα increases ATX production by Huh7, HepG2, and Hep3B liver cancer cells through activating nuclear factor κB (NFκB). The subsequent increase in LPA enhanced the invasiveness of the cancer cells [145]. The secretion of IL-8 is increased by LPA in human bronchial epithelial cells, which is mediated by protein kinase Cδ (PKCδ) and NFκB [146,147]. IL-8 and IL-6 expressions in ovarian cancer cells are also increased by LPAR2 or LPAR3 activation [148]. In a colon cancer model, LPAR2 knockout mice formed smaller tumors after induction with azoxymethane (AOM)/dextran sulfate sodium (DSS). This was accompanied by decreased levels of COX2 and CCL2 and reduced macrophage infiltration [149]. Zhao et al. showed that LPP1 inhibits LPA-induced NFκB translocation, which blocks IL-8 secretion in human bronchial epithelial cells [150]. This suggests an important role of LPP1 in inflammation [50,142].
We recently proposed a model of the ATX–LPA inflammatory cycle in breast cancer [151,152]. In this model, tumor-derived inflammatory cytokines such as TNFα and IL-1β increase ATX secretion by the adjacent mammary adipose. As a consequence, LPA levels increase in the TME. The increased LPA stimulates cancer cells to produce more cytokines, which can overcome the LPA-mediated feedback inhibition of mRNA expression for ATX [153] to form a feed-forward inflammatory cycle. This ATX–LPA inflammatory cycle can be exacerbated by radiotherapy (RT), since irradiation increases COX-2 and inflammatory cytokines in cultured adipose tissues as well as in the fat pads of mice [154,155]. ATX and LPAR1/2 levels are also elevated by irradiation. Dexamethasone, an anti-inflammatory glucocorticoid, attenuates the RT-induced upregulation of the expression ATX and LPA1R and LPA2R and increases LPP1 expression [156], which together decrease LPA signaling. Pulmonary fibrosis caused by RT or bleomycin is also blocked by dexamethasone [157,158,159].
It is well documented that LPA signaling promotes cell survival by inhibiting the intrinsic and extrinsic apoptosis pathways [160]. LPA decreases the level of the Fas receptor and reduces the expression of the Fas ligand [161,162], which makes cancer cells less responsive to the extrinsic pro-apoptotic stimuli. LPA also attenuates the intrinsic apoptosis pathway by increasing Bcl-2 and inhibiting Bad and Bax [163,164]. These effects of LPA depend on the activation of the PI3K–Akt pathway. The decrease in the sensitivity of cancer cells to chemotherapy and RT is contributed, at least partly, by the upregulation of LPA signaling. LPA decreases the effectiveness of Taxol [165], tamoxifen [166], and doxorubicin [167] in killing breast cancer cells.
The critical role of LPAR2 in protecting cells from radiation-induced damage has been illustrated by LPAR2 knockout mice, which exhibit increased irradiation-induced apoptosis in intestinal tissue [168]. By contrast, the knockout of LPAR1 or LPAR3 does not have this effect [168]. On the other hand, LPAR2 agonists show a therapeutic potential against irradiation-induced injury [168,169]. LPA contributes to the resistance of 786-O renal cancer cells to Temsirolimus and Sunitinib by activating Arf6 GTPase through LPAR2 [170]. Similarly, blocking LPAR1/3 with Ki16425 in resistant UMRC3 renal cancer cells re-establishes the sensitivity to Sunitinib [171]. The long-term culture of PANC-1 pancreatic cancer cells in the presence of cisplatin results in an upregulation of LPAR3 [172]. LPA through the activation of LPA1R and PI3K stabilized the expression of nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor, which through the anti-oxidant response element increases the expression of the multidrug-resistant transporters, anti-oxidant genes, and enzymes of DNA repair [166,167,173,174]. Thus, the ATX inhibitors, ONO-8430506 and GLPG1690, enhance the sensitivity of breast tumor to doxorubicin and RT [167,175]. It should be noted that the later effect of GLPG1690 involved decreased cell division in the cancer cells, and this is compatible with the major effect of RT in solid tumors being to increase cell senescence rather than apoptosis [176,177,178]. Effects of the upregulation of LPA signaling in cancer cells are summarized in Figure 3.
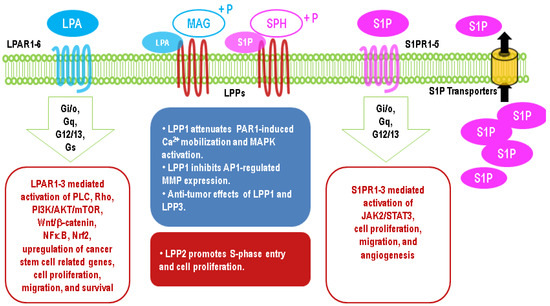
Figure 3. Major effects of upregulation of lysophosphatidate (LPA) and sphingosine 1-phosphate (S1P) signaling in cancer cells through G protein-coupled receptors and different functions of LPP1/3 and LPP2 in cancers.

