1. KRAS as a Pro-Inflammatory Modulator of the Tumor Microenvironment
KRAS mutations have generally been more related to an anti-inflammatory and, consequently, pro-tumor microenvironment rather than a pro-inflammatory one. However, several studies also reported the association of KRAS with pro-inflammatory/anti-tumor cytokines and chemokines. In fact,
KRAS mutations have been related to pro-inflammatory chemokines, such as ICAM-1, IL-18, and IL-6. Nevertheless, while the first two have been frequently associated with pro-inflammatory functions
[1][2] (
Figure 1), IL-6 has been described to exert an anti-inflammatory role in the
KRAS mutation context (
Figure 2)
[3].
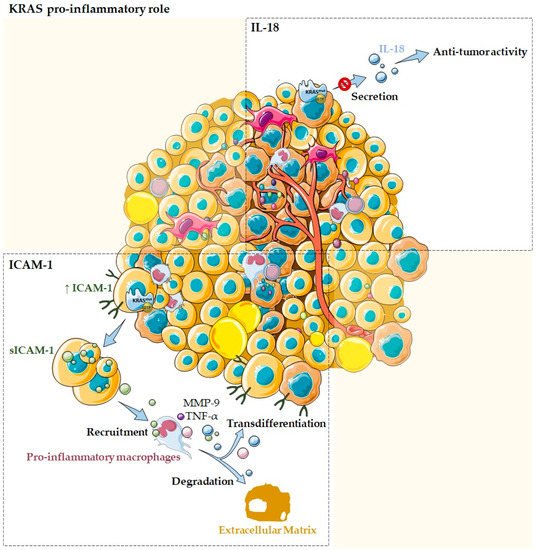
Figure 1. KRAS as a pro-inflammatory tumor microenvironment modulator. Several studies reported the association of KRAS with pro-inflammatory cytokines and chemokines, such as ICAM-1 and IL-18. Normal acinar cells transfected with oncogenic mutant KRAS are described to express high levels of ICAM-1, which is then secreted into its soluble form. The sICAM-1 acts as a chemoattractant for pro-inflammatory macrophages and stimulates them to produce MMP-9 that allow ECM degradation, as well as pro-inflammatory chemokines, such as TNF-α that can drive transdifferentiation signaling. KRAS mutations can also modulate the TME by impairing IL-18 secretion, blocking its immune-stimulatory function and, thus, contributing to evasion of the local immune system during tumor development.
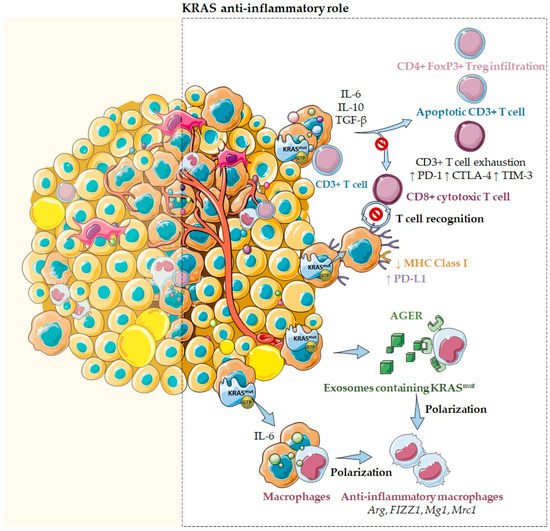
Figure 2. KRAS as an anti-inflammatory tumor microenvironment modulator. Several reports emphasize that KRAS mutations may sustain an anti-inflammatory microenvironment through the secretion of several inflammatory chemokines and cytokines, such as TGF-β, IL-10, and IL-6. In fact, cells harboring KRASG12D mutations secrete high levels of these anti-inflammatory mediators that inhibit T cell activation, suppress cytotoxic CD8+ T cell-mediated tumor killing, and convert pro-inflammatory CD4+ T cells to anti-inflammatory Tregs. Moreover, KRAS mutations were also described to induce the downregulation of MHC class I molecules and the upregulation of PD-L1, reducing the ability of CD8+ cytotoxic T cells to recognize and kill cancer cells. Additionally, KRAS mutations may drive an anti-inflammatory and pro-tumor immune suppressive microenvironment mediated through IL-6 secretion. Notably, when IL-6 was blocked, a reduction of anti-inflammatory macrophage gene expression, such as Arg, FIZZ1, Mg1, and Mrc1, and a reduction of the immunosuppressive cytokines TGF-β and IL-10 were observed. Moreover, it has also been described that IL-6 induces higher levels of T cell exhaustion markers, such as PD-1, CTLA-4, and TIM-3. Furthermore, KRAS mutations effects can also be mediated through exosomes containing KRASG12D. These exosomes can be taken via an AGER-dependent mechanism—a multiligand receptor—by macrophages, modulating their differentiation into a pro-tumor/anti-inflammatory phenotype.
In the pancreas, normal acinar cells transfected with oncogenic mutant
KRAS are described to express high levels of ICAM-1, which is then converted to a soluble form, the sICAM-1
[4]. In its turn, sICAM-1 acts as a chemoattractant for immune cells, namely, M1-like/pro-inflammatory macrophages, but not for M2-like/anti-inflammatory ones. Attracted pro-inflammatory macrophages directly interact with acinar cells via membrane ICAM-1, providing enzymes that allow ECM degradation, such as matrix metalloproteinase 9 (MMP-9), as well as inflammatory cytokines and chemokines that can drive transdifferentiation signaling, such as TNF-α (
Figure 1). This process is believed to contribute to acinar cells metaplasia and to drive the initiation of precancer lesions, which ultimately can progress to pancreatic cancer
[1][4][5]. Overall, these data support KRAS, ICAM-1, and inflammation as contributors to pancreatic ductal adenocarcinoma by initiation and acceleration of acinar-ductal metaplasia
[5].
KRAS mutations can also modulate the TME through IL-18, which is an immune-stimulatory cytokine
[6]. It is an important chemokine produced by epithelial cells of the gastrointestinal tract, the airway, and the skin and also by activated macrophages, Kupffer cells, B cells, and dendritic cells
[6]. IL-18 has been implicated in host immune defense against tumor development
[6]. Smakman and co-workers demonstrated, using the colorectal cancer cell line C26, that
KRAS knockdown resulted in the upregulation of IL-18 and its secretion into the medium
[1][6]. Authors also evidenced that C26 tumor growth in the liver can be strongly inhibited by the production of IL-18 by hepatocytes
[6]. Thus, this work demonstrated that
KRASG12D mutation suppresses IL-18 chemokine production, possibly contributing to evasion of the local immune system during tumor development (
Figure 1)
[1][6].
In lung cancer, to the best of our knowledge, there are no reports concerning the pro-inflammatory functions mediated by KRAS.
2. KRAS as an Anti-Inflammatory Modulator of the Tumor Microenvironment
Paradoxically, tumors harboring
KRAS mutations have also been associated with an immunosuppressive and anti-inflammatory microenvironment (
Figure 2). In colon cancer,
KRASG12V mutants are described to catalyze the differentiation of pro-inflammatory T cells into immunosuppressive T regulatory cells (Tregs) and promote their infiltration in a
KRAS-driven lung tumorigenesis mouse model
[7]. In lung cancer,
KRAS mutations are associated with high levels of Treg infiltration
[7], especially the
KRASG12D mutation, which induces CD3
+ T cell apoptosis and impairs the cytotoxic CD8
+ T cell activation
[7]. Additionally, in pancreatic cancer, cells harboring
KRASG12D mutations secrete high levels of the anti-inflammatory mediators TGF-β and IL10, crucial chemokines for sustaining an immunosuppressive environment and cancer cell immune escape
[7]. Among their multitude of functions, IL-10 is well-known to inhibit T cell activation, whereas TGF-β inhibits T cell activation and proliferation and promotes epithelial to mesenchymal transition, favoring cancer cell migration and invasion. Additionally, IL-10 and TGF-β released by pancreatic cancer cells are described to suppress cytotoxic CD8
+ T cell-mediated tumor killing
[7]. Moreover, in pancreatic cancer, it was also reported that
KRAS mutations effects could be mediated through exosomes. In fact, Dai and collaborators reported that exosomes containing
KRASG12D are released by dead, dying, or stressed cells, such as cancer cells
[8]. These exosomes can be taken via an AGER (advanced glycosylation end product-specific receptor) dependent mechanism—a multiligand receptor—by macrophages. This process causes their differentiation into an M2-like pro-tumor/anti-inflammatory phenotype through the signal transducer and activator of transcription 3 (STAT3)-dependent fatty acid oxidation mechanism (
Figure 2)
[8].
Other reports emphasize that
KRAS mutations may sustain an anti-inflammatory microenvironment through the secretion of several inflammatory chemokines and cytokines, such as IL-6, IL-10, and GM-CSF
[3].
In fact, high IL-6 secretion was observed in different cell types harboring oncogenic
KRAS mutations, such as human lung and kidney cells, fibroblasts, and myoblasts
[1]. In lung cancer cells,
KRAS mutations seem to promote increased levels of IL-6 via NF-kB, resulting in the activation of the STAT3 pathway
[3]. In its turn, IL-6-mediated STAT3 activation may contribute to immunosuppressive MDSCs accumulation
[3]. Paradoxically, IL-6 was also associated with pro-tumor Treg/Th17 cell response due to the observation that anti-IL-6 treatment promotes a T cell response switch, from a pro-tumor Treg/Th17 to an anti-tumor cytotoxic CD8
+ T cell response
[3]. Therefore, IL-6 may re-educate the lung microenvironment towards an anti-inflammatory phenotype, limiting inflammation via polarization of anti-inflammatory macrophages, recruitment of MDSCs and Treg/Th17 increasing response, favoring tumor immune escape and growth
[3]. In pancreatic cancer,
KRAS mutations are present in the majority of the cases, as are high levels of IL-6. In fact, Ras-driven pancreatic tumors are described to promote IL-6 secretion leading to STAT3 signaling pathway activation
[9][2]. Interestingly, both molecules are required as mediators of
KRAS mutations to promote pancreatic cancer precursor lesions initiation and progression to pancreatic ductal adenocarcinoma
[1]. Notably, when IL-6 is blocked, a reduction of anti-inflammatory macrophage gene expression, such as Arginase1 (
Arg), Found in inflammatory zone 1 (
FIZZ1), Macrophage galactose binding lectin (
Mg1), and Mannose receptor C type 1 (
Mrc1), and a reduction of the immunosuppressive cytokines TGF-β and IL-10 were observed. Moreover, a downregulation of the surface expression of the Natural killer group 2 member D receptor (NKG2D or CD314) was also described on NK cells as a mechanism to escape NK cell-mediated cytotoxicity in
KRAS-driven lung murine models
[10]. Additionally, although activated and effector-memory CD8
+ T cells are described to increase in
KRAS mutated mouse models, this seems not to be sufficient to impair tumor growth, suggesting the presence of parallel immune escape mechanisms
[10]. It has also been described that IL-6 stimulates the recruitment of neutrophils, decreases T-cell infiltration, and induces higher levels of T-cell exhaustion markers, such as programmed cell death 1 (PD-1), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3). Altogether, these data demonstrate that
KRAS mutations may drive an anti-inflammatory and pro-tumor immune suppressive microenvironment mediated through IL-6 secretion (
Figure 2).
Nevertheless, IL-10 is also modulated by
KRAS mutations. In fact, in colorectal cancer cells harboring
KRAS mutations, an upregulation of the anti-inflammatory cytokine IL-10 via the MEK/ERK/AP-1 pathway was observed. Secreted IL-10 enabled the conversion of pro-inflammatory CD4
+ T cells to anti-inflammatory CD4
+ FoxP3
+ Tregs cells
[1][2][11]. In addition, it was described that
KRASG12D could promote Treg transformation by blocking the interferon regulatory factor 2 (IRF2), resulting in repression of IRF2/CXCL3 pathway and binding of CXCL3 to CXCR2 on MDSCs, driving immune suppression and immune therapy resistance in colorectal cancer
[1][11]. Similarly, in pancreatic cancer, it was also confirmed that IL-10 stimulates the conversion of CD4
+ T cells to CD4
+ FoxP3
+ Tregs cells
[1][2][11]. In lung cancer, IL-10 was also reported to mediate the recruitment of anti-inflammatory M2 macrophages and Tregs to the tumor
[2].
Interestingly, GM-CSF was also identified as a transcriptional target of oncogenic
KRAS in pancreatic ductal epithelial cells and in colorectal cancer
[1][12][13]. GM-CSF serves as a proliferation and maturation factor of several myeloid cells and has the potential to promote both anti- and pro-inflammatory effects
[14]. In pancreatic cancer, GM-CSF is produced in response to activation of KRAS through the concerted action of multiple effectors, such as ERK and PI3K
[14]. Additionally, it is related to the expansion of immunosuppressive Gr1
+ CD11b
+ myeloid cells. However, GM-CSF is not unique in this ability; IL1-β, IL-6, and VEGF also have this capacity and, curiously, are also targets of oncogenic Ras signaling
[14].
Importantly,
KRAS mutations were also described to induce the downregulation of major histocompatibility complex (MHC) class I molecules, reducing the ability of CD8
+ cytotoxic T cells to recognize and kill cancer cells (
Figure 2)
[1].
In addition,
KRAS mutation status has correlated positively with the programmed death-ligand 1 (PD-L1) expression in distinct cancers
[1][15]. In
KRAS mutant lung cancer cells, oncogenic
KRAS was proven to upregulate PD-L1 through an increase in PD-L1 mRNA stability mediated by the AU-rich element-binding protein tristetraprolin (TTP)
[1]. This expression is regulated by MAPK-dependent transcriptional activity of the activator protein 1 (AP-1) and by STAT3
[1]. More recently, a correlation between
KRAS mutations, increased PD-L1 expression, and increased CD8
+ tumor-infiltrating lymphocytes was observed, linking
KRAS mutations as a promoter of an anti-inflammatory, immunosuppressive TME, adaptive immune resistance, and tumor immunogenicity (
Figure 2)
[1][15]. Other relevant studies demonstrated that the co-mutation of
TP53 and
KRAS led to an immune-rich microenvironment of high tumor mutation burden (TMB), enhanced PD-L1 expression, and enrichment of immune cell infiltration, namely, CD4 memory T cells, NK cells, and M1 macrophages. In lung adenocarcinoma, the
TP53/
KRAS co-mutation induced an increased expression of PD-L1
[16]. These co-mutations were also reported to play a role in the activation of immune escape and anti-tumor immunity
[17][18]. In fact, it was reported that KRAS and TP53 cooperate to promote tumor and immune invasion by activating the ARF6/AMAp1 pathway, which provokes PD-L1 recycling and its cell surface expression
[1]. The induction of an immunosuppressive microenvironment orchestrated by
KRAS mutants seems to be also dependent on the transcription regulator Yap, due to the observation that Yap ablation in
KRAS/
TP53 mutant pancreatic cells prevents MDSC recruitment favoring MHCII
+ anti-tumor macrophages, resulting in T cell reactivation, apoptosis of neoplastic cells, and tissue regeneration
[18]. In detail, Yap binds to the promoter region of CSFS and of IL-6, controlling their transcription in
KRAS/
TP53 mutant pancreatic cells. In the absence of Yap, IL-6 and CSFS are blocked, and INF-γ, IL-12, IL-15, IL-4, and IL-13 are produced, stimulating T cell activity
[18]. Finally,
KRAS and
MYC also cooperate to establish an immune-suppressive stroma through the involvement of CCL9 mediated recruitment of macrophages, PD-L1 and IL-23 dependent exclusion of T and B cells and natural killer (NK) cells
[19]. Overall,
KRAS mutations have a more relevant impact on promoting an anti-inflammatory microenvironment beneficial for tumorigenesis and immune escape than the opposite. The impact of such effects on tumor immune escape and progression is evident, but their contribution to the success of therapeutic response should be further exploited in the near future.
 +1 credit
+1 credit