 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Masayuki Yamamoto | + 5120 word(s) | 5120 | 2022-02-18 07:49:50 | | | |
| 2 | Vicky Zhou | -2085 word(s) | 3035 | 2022-02-22 03:33:40 | | |
Video Upload Options
Skin is constantly exposed to environmental insults, including toxic chemicals and oxidative stress. These insults often provoke perturbation of epidermal homeostasis and lead to characteristic skin diseases. AHR (aryl hydrocarbon receptor) and NRF2 (nuclear factor erythroid 2-related factor 2) are transcription factors that induce a battery of cytoprotective genes encoding detoxication and antioxidant enzymes in response to environmental insults. In addition to their basic functions as key regulators of xenobiotic and oxidant detoxification, it has been revealed that AHR and NRF2 also play critical roles in the maintenance of skin homeostasis. In fact, specific disruption of AHR function in the skin has been found to be associated with the pathogenesis of various skin diseases, most prevalently atopic dermatitis (AD).
1. Roles of AHR and NRF2 in Xenobiotic Detoxification
Transcription factor AHR (aryl hydrocarbon receptor) is a ligand-activated transcription factor that belongs to a basic helix-loop-helix/PER-ARNT-SIM (bHLH-PAS) family [1]. AHR is activated by the binding of a large variety of exogenous ligands, which include environmental pollutants such as polycyclic aromatic hydrocarbons (PAHs) and dioxins (Figure 1a). In the absence of a ligand, AHR forms a complex in the cytoplasm with at least three factors: Heat shock protein 90 (HSP90), the cochaperone protein p23 and hepatitis B virus X-associated protein 2 (XAP2) [2]. Upon binding of certain ligands to AHR, the AHR-HSP90-p23-XAP2 complex is disrupted, leading to the nuclear translocation of AHR. In the nucleus, AHR dimerizes with ARNT (AHR nuclear translocator) [3]. The AHR-ARNT heterodimer binds to XRE (xenobiotic response element) in regulatory regions of various AHR-target genes [4]. Prototype AHR-target genes include phase I detoxification enzymes such as cytochrome P450 (CYPs). Two AHR regulatory mechanisms have been reported. One is proteasomal degradation of the AHR protein, while the other is negative feedback regulation of AHR activation by the AHR repressor (AHRR) [5]. AHRR is known to be an AHR-target gene, and AHRR inhibits AHR activity by competing with AHR for dimerization with ARNT.
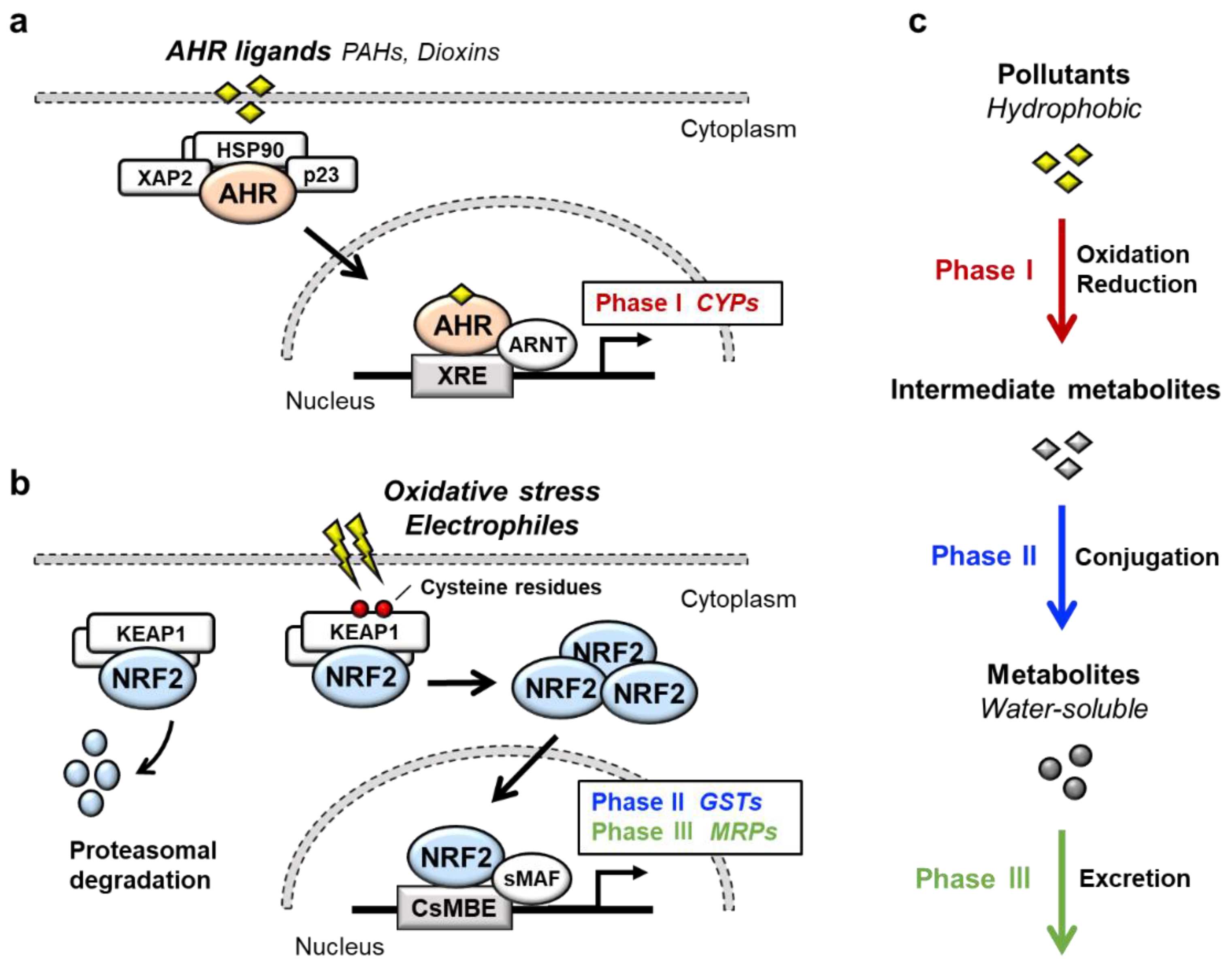
NRF2 (nuclear factor erythroid 2-related factor 2) is a transcription factor belonging to the cap‘n’collar (CNC) family. NRF2 possesses a basic leucine zipper (bZIP) and a CNC structure. NRF2 induces a battery of cytoprotective genes in response to oxidative and electrophilic stresses [6][7]. Under physiological conditions, NRF2 is bound by the repressor protein KEAP1 (kelch-like ECH-associated protein 1) in the cytoplasm, which promotes NRF2 degradation by the ubiquitin–proteasome pathway (Figure 1b) [8][9]. Upon exposure to oxidative or electrophilic stresses, specific reactive cysteine residues of KEAP1 are modified, which leads to the disruption of the KEAP1-NRF2 interaction [10][11]. This stabilizes NRF2 so that NRF2 translocates to the nucleus and forms a heterodimer with small MAF (sMAF) protein. The NRF2-sMAF heterodimer complex binds to the CNC-sMAF binding element (CsMBE), which is also known as the antioxidant response element (ARE) or electrophile response element (EpRE), in the regulatory regions of target genes [12][13]. Closer examination of the NRF2-target genes revealed that a set of genes encoding phase II detoxification enzymes such as glutathione S-transferase (GSTs), antioxidative enzymes such as NAD(P)H:quinone oxidoreductase 1 (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC) and phase III transporters such as multidrug-resistance-associated proteins (MRPs) in a member of ATP-binding cassette (ABC) transporters are the target genes of NRF2.
AHR and NRF2 are the key regulators of cytoprotective responses to environmental stresses. Both transcription factors play important roles in the transformation of hydrophobic molecules to water-soluble molecules that are easily eliminated from the body via urine, stool and sweat. Xenobiotic detoxification consists of three phases [14] (Figure 1c); the phase I detoxification reaction involves oxidation or reduction of xenobiotics, resulting in the conversion to more polar intermediate metabolites such as electrophiles. The phase II detoxification reaction involves conjugation of a hydrophilic moiety, e.g., glucuronate, sulfate, glutathione or glycine, to phase I metabolites. This reaction is catalyzed by a group of enzymes called transferases, and phase I metabolites are transformed into water-soluble molecules by transferases. In the phase III reaction, the conjugated metabolites are eliminated from the cells by transporters. It has been shown that AHR regulates the expression of phase I enzymes, such as CYP1A1, CYP1A2 and CYP1B1, while NRF2 regulates phase II enzymes, such as GSTA1, GSTP1 and UDP-glucuronosyl transferases (UGTs), and phase III transporters, such as MRPs.
2. Role of AHR in Epidermal Homeostasis
A well-known function of AHR is the regulation of xenobiotic metabolism through the induction of CYPs. However, many lines of evidence have reported that ligand-activated AHR also acts to induce the expression of genes involved in skin barrier formation [15][16][17][18]. In fact, exposure of normal human epidermal keratinocytes to TCDD increases the mRNA expression of 40% (24/60) of the epidermal differentiation complex (EDC) genes involved in cornified cell envelope formation, such as FLG, FLG2, LCE1C, LCE2A, LCE2B, LCE3A, LCE3E, SPRR1A, SPRR2A, SPRR2B, S100A9, S100A12, S100A7, Repetin (RPTN) and Hornerin (HRNR) [17][18]. In organotypic cultures of human keratinocytes, treatment with TCDD causes well-developed SC, indicating that TCDD accelerates the onset of epidermal terminal differentiation [15]. Additionally, exposure to TCDD accelerates the formation of the fetal mouse skin barrier in utero [17]. Conversely, in primary keratinocytes derived from wild-type mice, the expression of terminal differentiation genes is suppressed by treatment with an AhR antagonist GNF351 or a selective AhR modulator SGA360 [19]. The expression of terminal differentiation genes is reproducibly repressed in AhR-knockout mouse primary keratinocytes [19]. These results support the physiological roles of AhR during epidermal differentiation.
AhR-knockout mice were generated in 1996-1997 independently in three distinct laboratories [20][21][22]. Fernandez-Salguero et al. reported an AhR-knockout mouse line generated by replacing a part of the first exon of the AhR gene with a neomycin resistance cassette (NEO). The mice revealed severe hyperkeratosis, acanthosis and marked dermal fibrosis in the dorsal skin [20]. In contrast, the other two lines of AhR-knockout mice did not show characteristic skin phenotypes. The second and third knockout mouse lines were generated by the replacement of a part of the second exon with NEO and LacZ fused to a nuclear localization sequence, respectively [21][22]. While these differences in gene targeting strategy may contribute to the phenotypic difference of the skin, the precise reason for the phenotypic difference is currently unclear.
Consistent with the results of the latter two mouse lines, keratinocyte-specific AhR-knockout mice using Keratin 14 (K14)-Cre (AhRflox::K14-Cre mice) show no obvious macroscopic phenotypes in unstressed skin [23]. However, when the upper layers of SC are mechanically removed by tape stripping, transepidermal water loss (TEWL) values in AhRflox::K14-Cre mice are substantially increased compared with those in control mice, suggesting that AhR plays a role in skin barrier function [23]. These studies using AhR-knockout cell lines and AhR-knockout mice support the notion that AHR is involved in epidermal homeostasis [19][20], although there remain many unsolved questions.
ARNT is indispensable for the transcriptional activity of AHR. Keratinocyte-specific Arnt-knockout mice using Keratin 5 (K5)-Cre (Arnt flox::K5-Cre mice) show severe impairment of the epidermal barrier [24]. Arnt flox::K5-Cre mice exhibit a loss of body weight and die within 24 h after birth [24]. TEWL is increased in the epidermis of Arnt flox::K5-Cre mice, and the application of salve to the skin retards weight loss; the mice survive longer than 24 h [24]. These observations indicate that skin barrier dysfunction results in severe dehydration in Arnt flox::K5-Cre mice.
Consistent with the results of the Arnt flox::K5-Cre mice, keratinocyte-specific Arnt-knockout mice using K14-Cre (Arnt flox::K14-Cre mice) also die with a failure of epidermal barrier function [25]. Microarray analyses of the epidermis of Arnt flox::K14-Cre mice revealed upregulation of several EDC genes, including S100a genes (S100a8, S100a9, S100a10) and Sprrs (Sprr1a, Sprr2i, Sprr2j, Sprrl1) [25]. Furthermore, serine protease inhibitors, including secretory leukocyte protease inhibitor (Slpi), are also upregulated and are involved in SC desquamation [25]. While Hif1α (hypoxia-inducible factor 1α) can also form a heterodimer with Arnt, keratinocyte-specific Hif1α-knockout mice using K5-Cre (Hif1αflox::K5-Cre mice) show no obvious phenotype, suggesting that the AhR-Arnt heterodimer rather than Hif1α-Arnt is responsible for epidermal homeostasis [24]. These wide-ranging reports suggest that AhR or Arnt deficiency impairs the development and function of the epidermal barrier, but further clarifications are required to understand the general features.
3. Role of NRF2 in Epidermal Homeostasis
Showing very good agreement with the results of Keap1-knockout mice, a transgenic mouse line expressing a constitutively active Nrf2 mutant (caNrf2) specifically in keratinocytes was shown to display scaling and dry skin [27]. caNrf2 lacks the Neh2 domain, which is responsible for binding to Keap1, and this molecule is expressed under the control of a strong CMV enhancer and specific removal of the stopper cassette by K5-Cre (caNrf2::K5-Cre mouse). Histological analyses of the caNrf2 transgenic mice showed thickening of the epithelium (acanthosis) and severe hyperkeratosis in the skin. Microarray analyses using the skin of caNrf2::K5-Cre mice at postnatal Day 2.5 revealed upregulation of genes involved in epidermal barrier formation, including Slpi, Sprr2d and Sprr2h [27]. These mouse phenotypes suggest that prolonged activation of Nrf2 in keratinocytes disturbs skin homeostasis.
Supporting this notion, this research group has also reported that Nrf2 activation in newborn mice induced by topical application with sulforaphane or tert-butyl hydroquinone (tBHQ) for 10 days also causes skin abnormalities resembling those of caNrf2::K5-Cre mice [27]. However, the concentrations of the inducers applied were 10 mM and 50 mM for sulforaphane and tBHQ, respectively, which are very high doses. Therefore, the relationship between pharmacological induction of skin phenotypes and Nrf2 contribution awaits further verification.
In this regard, crossing a transgenic mouse line expressing the dominant-negative form of Nrf2 (dnNrf2) under the control of the Lor promoter to Lor-knockout mice (Lor-knockout::dnNrf2 mice) showed a distinct phenotype. Inhibition of endogenous Nrf2 activity by dnNrf2 in the Lor-deficient epidermis causes severe loss of barrier function and death within 24 h [28]. LOR is the main component of the cornified cell envelope. While Lor-knockout mice show a delay in the formation of the epidermal barrier in utero, the mice can survive with the formation of a functional barrier by birth [29]. Other cornified cell envelope components, including Sprrs and Lce1, are upregulated in the epidermis of Lor-knockout mice, so they might compensate for the loss of Lor [29][30]. In fact, the expression of Sprrs and Lce1 genes is decreased to basal levels in the epidermis of Lor-knockout::dnNrf2 mice [28][30]. ChIP assays and reporter assays show that Nrf2 directly upregulates the Sprrs (Sprr2d and Sprr2h) and Lce1 genes (Lce1b, Lce1c, Lce1e, Lce1g, Lce1h and Lce1m) [28][30]. These results suggest that Nrf2 activation in Lor-knockout mice may act to induce a compensatory response to repair the defective barrier. While the Keap1-Nrf2 system is generally the major cellular protection mechanism against xenobiotic and oxidative stresses, these reports suggest that Nrf2 plays important roles in the maintenance of skin homeostasis, similar to AhR.
4. AHR as Therapeutic Target for Atopic Dermatitis
Atopic dermatitis (AD) is a chronic recurrent inflammatory skin disease characterized by epidermal barrier dysfunction and immune dysregulation. Loss-of-function variants in FLG, which is essential for the formation of the epidermal barrier, have been reported to be the most common risk factor for the development of AD [31]. However, only 40% of individuals with FLG variations develop AD symptoms [31][32][33]. These facts indicate that other factors are also involved in the pathogenesis of AD [33][34].
It has been shown that AD patients account for up to 25% of children and 2–3% of adults [35]. Of note, the international study of asthma and allergies in childhood (ISAAC), which is a worldwide epidemiological research in more than 50 countries, revealed that the prevalence of AD increased among schoolchildren between ISAAC phase I (1992-1996) and phase III (2000-2003) studies in many parts of the world [36]. In addition, a systematic review including 69 reports showed that the prevalence of AD between 1990 and 2010 increased in Africa, East Asia and parts of Europe [37]. While the pathophysiology of AD appears to be multifactorial with complex interactions between genetic and environmental factors, the recent increase in the prevalence of AD supports the attribution of environmental factors in predisposed individuals [38].
In fact, several lines of epidemiological research provide evidence that exposure of the skin to air pollutants acts as a risk factor for the development or aggravation of AD. Outdoor air pollutants are mostly generated from the burning of fossil fuels for vehicles and industries [39], and components of air pollutants mainly include particulate matter (PM), which is a mixture of solid or liquid particles suspended in the air. Outdoor concentrations of PM are associated with increased AD symptoms and itching [40][41]. A meta-analysis of 13 studies showed positive correlations between PM exposure and AD [42]. Importantly, PM contains PAHs, prototypical AHR ligands, and induces CYP1A1 mRNA expression in an AHR-dependent manner in human primary keratinocytes [43]. In addition to PM, active smoking and passive exposure to tobacco in the home are also associated with a higher prevalence of AD in both children and adults [44]. Tobacco smoke is the major source of indoor pollutants and contains numerous chemical constituents, including PAHs [45]. These reports support the hypothesis that AHR activation is involved in AD caused by air pollutants.
A model study employing constitutively active AhR in mice supported the hypothesis that AhR plays an important role in the development of AD [46]. To analyze the effects of chronic AhR activation in the epidermis, a transgenic mouse expressing a constitutively active form of AhR was generated under the control of the K14 promoter (AhR-CA mice). AhR-CA mice develop erosive eczema in the face and back skin with highly frequent scratching [46]. Histological examination shows acanthosis, hyperkeratosis and abundant infiltration of cells related to Th2-type inflammation in the skin [47]. These phenotypes highly recapitulate those of patients with AD, indicating that the constitutive activation of AhR provokes pathological conditions similar to those frequently observed in patients with AD.
Notably, in the epidermis of AhR-CA mice, the neurotrophic factor Artemin is highly expressed, which causes extension of sensory nerves into the epidermis and provokes hypersensitivity to itch or alloknesis [47][48]. The Artemin gene is directly regulated by AhR via an XRE-containing enhancer located 52 kb upstream of the gene [49]. Showing very good agreement with these results, in the skin of AD patients, AHR is activated, followed by upregulation of the mRNA and protein levels of CYP1A1 and ARTEMIN [47][50].
In contrast, it has been reported that AHR activation improves skin barrier function by accelerating epidermal terminal differentiation. For instance, coal tar has been used as a therapeutic agent for inflammatory skin diseases, including AD. In fact, topical application of coal tar restores the expression of skin barrier proteins, including FLG, in the skin of patients with AD [51]. Coal tar contains abundant PAHs and has been shown to induce epidermal differentiation via AHR. The efficacy of coal tar seems to be attributable to AHR activation [51].
In addition, tapinarof, 3,5-dihydroxy-4-isopropyl-trans-stilbene, a natural origin small molecule, shows efficacy in patients with AD and is now in clinical development for the treatment of skin diseases. In a phase 2 double-blind study, AD symptom was improved in the patients treated with tapinarof cream [52][53]. Recent research revealed that tapinarof activates AHR through direct binding as an agonist and induces mRNA expression of epidermal terminal differentiation markers, including FLG and IVL, in primary human keratinocytes [54]. Imiquimod has been used to provoke skin inflammation that mimics psoriasis, and tapinarof reduces imiquimod-induced inflammation in mouse skin in an AhR-dependent manner [54]. Similar to the case for coal tar, the efficacy of tapinarof is considered to depend, at least partially, on the AhR activity. These wide-ranging observations thus demonstrate that AHR activation acts to repair skin barrier function, but in turn, excessive expression of AHR leads to the development of AD (Figure 2). These reports demonstrated that AHR is a potential therapeutic target, and moderate-level activation of AhR may have therapeutic efficacy for skin barrier recovery. In contrast, AHR inhibitors seem to be important for the treatment of AD.
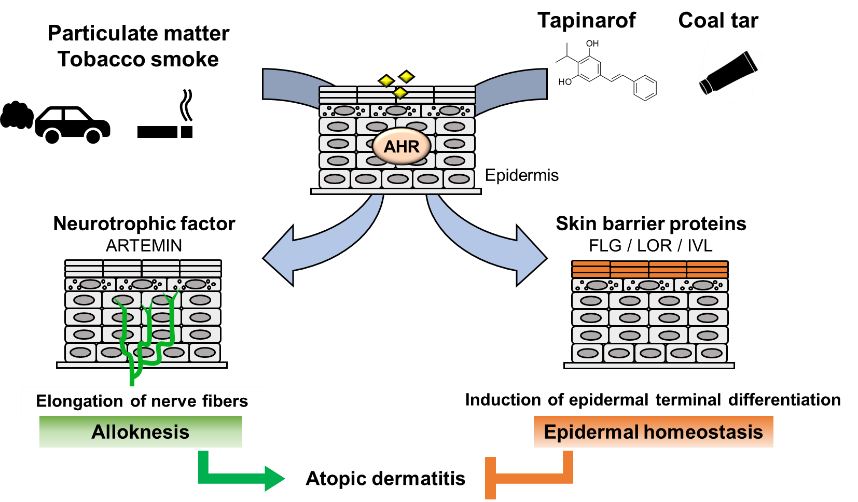
Figure 2. Dual functions of AHR in atopic dermatitis. PM and tobacco smoke contain PAHs that activate AHR. Activation of AHR induces the expression of the neurotrophic factor Artemin. Upregulation of Artemin causes extension of cutaneous sensory nerves into the epidermis and provokes hypersensitivity to itch or alloknesis. Tapinarof and coal tar improve skin barrier function by accelerating epidermal terminal differentiation.
References
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Commun 1992, 184, 246-253, doi:10.1016/0006-291x(92)91185-s.
- Petrulis, J.R.; Perdew, G.H. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem Biol Interact 2002, 20, 25-40, doi:10.1016/s0009-2797(02)00064-9.
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the ah receptor nuclear translocator protein (arnt) as a component of the DNA binding form of the ah receptor. Science 1992, 256, 1193-1195, doi:10.1126/science.256.5060.1193.
- Fujisawa-Sehara, A.; Yamane, M.; Fujii-Kuriyama, Y. A DNA-binding factor specific for xenobiotic responsive elements of P-450c gene exists as a cryptic form in cytoplasm: its possible translocation to nucleus. Proc Natl Acad Sci U S A 1988, 85, 5859-5863, doi:10.1073/pnas.85.16.5859.
- Fujii-Kuriyama, Y.; Kawajiri, K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci 2010, 86, 40-53, doi:10.2183/pjab.86.40.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018, 98, 1169-1203, doi:10.1152/physrev.00023.2017.
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system as a molecular target of cancer treatment. Cancers (Basel) 2020, 13, doi:10.3390/cancers13010046.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999, 13, 76-86, doi:10.1101/gad.13.1.76.
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 2004, 24, 7130-7139, doi:10.1128/MCB.24.16.7130-7139.2004.
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol 2020, 40, e00099-00020, doi:10.1128/MCB.00099-20.
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol Cell Biol 2016, 36, 271-284, doi:10.1128/MCB.00868-15.
- Otsuki, A.; Yamamoto, M. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch Pharm Res 2020, 43, 275-285, doi:10.1007/s12272-019-01193-2.
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol 2005, 25, 8044-8051, doi:10.1128/MCB.25.18.8044-8051.2005.
- Kohle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem Pharmacol 2007, 73, 1853-1862, doi:10.1016/j.bcp.2007.01.009.
- Loertscher, J.A.; Sattler, C.A.; Allen-Hoffmann, B.L. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol Appl Pharmacol 2001, 175, 121-129, doi:10.1006/taap.2001.9202.
- Geusau, A.; Khorchide, M.; Mildner, M.; Pammer, J.; Eckhart, L.; Tschachler, E. 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs differentiation of normal human epidermal keratinocytes in a skin equivalent model. J Invest Dermatol 2005, 124, 275-277, doi:10.1111/j.0022-202X.2004.23541.x.
- Sutter, C.H.; Bodreddigari, S.; Campion, C.; Wible, R.S.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol Sci 2011, 124, 128-137, doi:10.1093/toxsci/kfr205.
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol Sci 2013, 132, 235-249, doi:10.1093/toxsci/kfs325.
- van den Bogaard, E.H.; Podolsky, M.A.; Smits, J.P.; Cui, X.; John, C.; Gowda, K.; Desai, D.; Amin, S.G.; Schalkwijk, J.; Perdew, G.H.; et al. Genetic and pharmacological analysis identifies a physiological role for the AHR in epidermal differentiation. J Invest Dermatol 2015, 135, 1320-1328, doi:10.1038/jid.2015.6.
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol 1997, 34, 605-614, doi:10.1177/030098589703400609.
- Mimura, J.; Yamashita, K.; Nakamura, K.; Morita, M.; Takagi, T.N.; Nakao, K.; Ema, M.; Sogawa, K.; Yasuda, M.; Katsuki, M.; et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) recepto. Genes Cells 1997, 2, 645-654, doi:10.1046/j.1365-2443.1997.1490345.x.
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 1996, 93, 6731-6736, doi:10.1073/pnas.93.13.6731.
- Haas, K.; Weighardt, H.; Deenen, R.; Kohrer, K.; Clausen, B.; Zahner, S.; Boukamp, P.; Bloch, W.; Krutmann, J.; Esser, C. Aryl hydrocarbon receptor in keratinocytes is essential for murine skin barrier integrity. J Invest Dermatol 2016, 136, 2260-2269, doi:10.1016/j.jid.2016.06.627.
- Takagi, S.; Tojo, H.; Tomita, S.; Sano, S.; Itami, S.; Hara, M.; Inoue, S.; Horie, K.; Kondoh, G.; Hosokawa, K.; et al. Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function. J Clin Invest 2003, 112, 1372-1382, doi:10.1172/jci200318513.
- Geng, S.; Mezentsev, A.; Kalachikov, S.; Raith, K.; Roop, D.R.; Panteleyev, A.A. Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J Cell Sci 2006, 119, 4901-4912, doi:10.1242/jcs.03282.
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 2003, 35, 238-245, doi:10.1038/ng1248.
- Schäfer, M.; Farwanah, H.; Willrodt, A.H.; Huebner, A.J.; Sandhoff, K.; Roop, D.; Hohl, D.; Bloch, W.; Werner, S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol Med 2012, 4, 364-379, doi:10.1002/emmm.201200219.
- Huebner, A.J.; Dai, D.; Morasso, M.; Schmidt, E.E.; Schafer, M.; Werner, S.; Roop, D.R. Amniotic fluid activates the nrf2/keap1 pathway to repair an epidermal barrier defect in utero. Dev Cell 2012, 23, 1238-1246, doi:10.1016/j.devcel.2012.11.002.
- Koch, P.J.; de Viragh, P.A.; Scharer, E.; Bundman, D.; Longley, M.A.; Bickenbach, J.; Kawachi, Y.; Suga, Y.; Zhou, Z.; Huber, M.; et al. Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J Cell Biochem 2000, 151, 389-400, doi:10.1083/jcb.151.2.389.
- Ishitsuka, Y.; Huebner, A.J.; Rice, R.H.; Koch, P.J.; Speransky, V.V.; Steven, A.C.; Roop, D.R. Lce1 family members are Nrf2-target genes that are induced to compensate for the loss of loricrin. J Invest Dermatol 2016, 136, 1656-1663, doi:10.1016/j.jid.2016.04.022.
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006, 38, 441-446, doi:10.1038/ng1767.
- Henderson, J.; Northstone, K.; Lee, S.P.; Liao, H.; Zhao, Y.; Pembrey, M.; Mukhopadhyay, S.; Smith, G.D.; Palmer, C.N.; McLean, W.H.; et al. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. J Allergy Clin Immunol 2008, 121, 872-877 e879, doi:10.1016/j.jaci.2008.01.026.
- Irvine, A.D.; McLean, W.H.I.; Leung, D.Y.M. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011, 365, 1315-1327, doi:10.1056/NEJMra1011040.
- Weidinger, S.; O'Sullivan, M.; Illig, T.; Baurecht, H.; Depner, M.; Rodriguez, E.; Ruether, A.; Klopp, N.; Vogelberg, C.; Weiland, S.K.; et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol 2008, 121, 1203-1209 e1201, doi:10.1016/j.jaci.2008.02.014.
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol 2014, 70, 338-351, doi:10.1016/j.jaad.2013.10.010.
- Nutten, S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab 2015, 66 Suppl 1, 8-16, doi:10.1159/000370220.
- Deckers, I.A.; McLean, S.; Linssen, S.; Mommers, M.; van Schayck, C.P.; Sheikh, A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS One 2012, 7, e39803, doi:10.1371/journal.pone.0039803.
- Stander, S. Atopic dermatitis. N Engl J Med 2021, 384, 1136-1143, doi:10.1056/NEJMra2023911.
- Roberts, W. Air pollution and skin disorders. Int J Womens Dermatol 2021, 7, 91-97, doi:10.1016/j.ijwd.2020.11.001.
- Kim, J.; Kim, E.H.; Oh, I.; Jung, K.; Han, Y.; Cheong, H.K.; Ahn, K. Symptoms of atopic dermatitis are influenced by outdoor air pollution. J Allergy Clin Immunol 2013, 132, 495-498 e491, doi:10.1016/j.jaci.2013.04.019.
- Song, S.; Lee, K.; Lee, Y.M.; Lee, J.H.; Lee, S.I.; Yu, S.D.; Paek, D. Acute health effects of urban fine and ultrafine particles on children with atopic dermatitis. Environ Res 2011, 111, 394-399, doi:10.1016/j.envres.2010.10.010.
- Ngoc, L.T.N.; Park, D.; Lee, Y.; Lee, Y.C. Systematic review and meta-analysis of human skin diseases due to particulate matter. Int J Environ Res Public Health 2017, 14, doi:10.3390/ijerph14121458.
- Kim, B.E.; Kim, J.; Goleva, E.; Berdyshev, E.; Lee, J.; Vang, K.A.; Lee, U.H.; Han, S.; Leung, S.; Hall, C.F.; et al. Particulate matter causes skin barrier dysfunction. JCI Insight 2021, 6, e145185, doi:10.1172/jci.insight.145185.
- Mueller, D.; Uibel, S.; Braun, M.; Klingelhoefer, D.; Takemura, M.; Groneberg, D.A. Tobacco smoke particles and indoor air quality (ToPIQ) - the protocol of a new study. J Occup Med Toxicol 2011, 6, 35, doi:10.1186/1745-6673-6-35.
- Egawa, M.; Kohno, Y.; Kumano, Y. Oxidative effects of cigarette smoke on the human skin. Int J Cosmet Sci 1998, 21, 83-98, doi:10.1046/j.1467-2494.1999.181656.x.
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol 2005, 25, 9360-9368, doi:10.1128/MCB.25.21.9360-9368.2005.
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat Immunol 2017, 18, 64-73, doi:10.1038/ni.3614.
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat Immunol 2020, 21, 1486-1495, doi:10.1038/s41590-020-0802-6.
- Edamitsu, T.; Taguchi, K.; Kobayashi, E.H.; Okuyama, R.; Yamamoto, M. Aryl hydrocarbon receptor directly regulates artemin gene expression. Mol Cell Biol 2019, 39, e00190-00119, doi:10.1128/mcb.00190-19.
- Kim, H.O.; Kim, J.H.; Chung, B.Y.; Choi, M.G.; Park, C.W. Increased expression of the aryl hydrocarbon receptor in patients with chronic inflammatory skin diseases. Exp Dermatol 2014, 23, 278-281, doi:10.1111/exd.12350.
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schroder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest 2013, 123, 917-927, doi:10.1172/JCI65642.
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J Am Acad Dermatol 2019, 80, 89-98 e83, doi:10.1016/j.jaad.2018.06.047.
- Paller, A.S.; Stein Gold, L.; Soung, J.; Tallman, A.M.; Rubenstein, D.S.; Gooderham, M. Efficacy and patient-reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J Am Acad Dermatol 2021, 84, 632-638, doi:10.1016/j.jaad.2020.05.135.
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest Dermatol 2017, 137, 2110-2119, doi:10.1016/j.jid.2017.05.004.


