 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gabriel Mbalaviele | + 1522 word(s) | 1522 | 2022-01-25 09:12:36 | | | |
| 2 | Jessie Wu | Meta information modification | 1522 | 2022-02-21 02:00:08 | | | | |
| 3 | Jessie Wu | + 36 word(s) | 1558 | 2022-02-21 02:03:24 | | | | |
| 4 | Jessie Wu | + 34 word(s) | 1556 | 2022-02-21 02:06:14 | | |
Video Upload Options
Adenosine diphosphate (ADP)-ribosylation is the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) to acceptor proteins. This post-translational modification (PTM) unavoidably alters protein functions and signaling networks, thereby impacting cell behaviors and tissue outcomes. As a ubiquitous mechanism, ADP-ribosylation affects multiple tissues, including bones, as abnormal ADP-ribosylation compromises bone development and remodeling.
1. Introduction
The transfer of adenosine diphosphate (ADP)-ribose unit(s) from nicotinamide adenine dinucleotide (NAD+) to acceptor proteins is known as ADP-ribosylation. This post-translational modification (PTM) unavoidably alters protein functions and signaling networks, thereby impacting cell behaviors and tissue outcomes. As a ubiquitous mechanism, ADP-ribosylation affects multiple tissues, including bones, as abnormal ADP-ribosylation compromises bone development and remodeling.
2. Osteoclast Differentiation
2.1. Role of ARTD1 in Osteoclast Differentiation
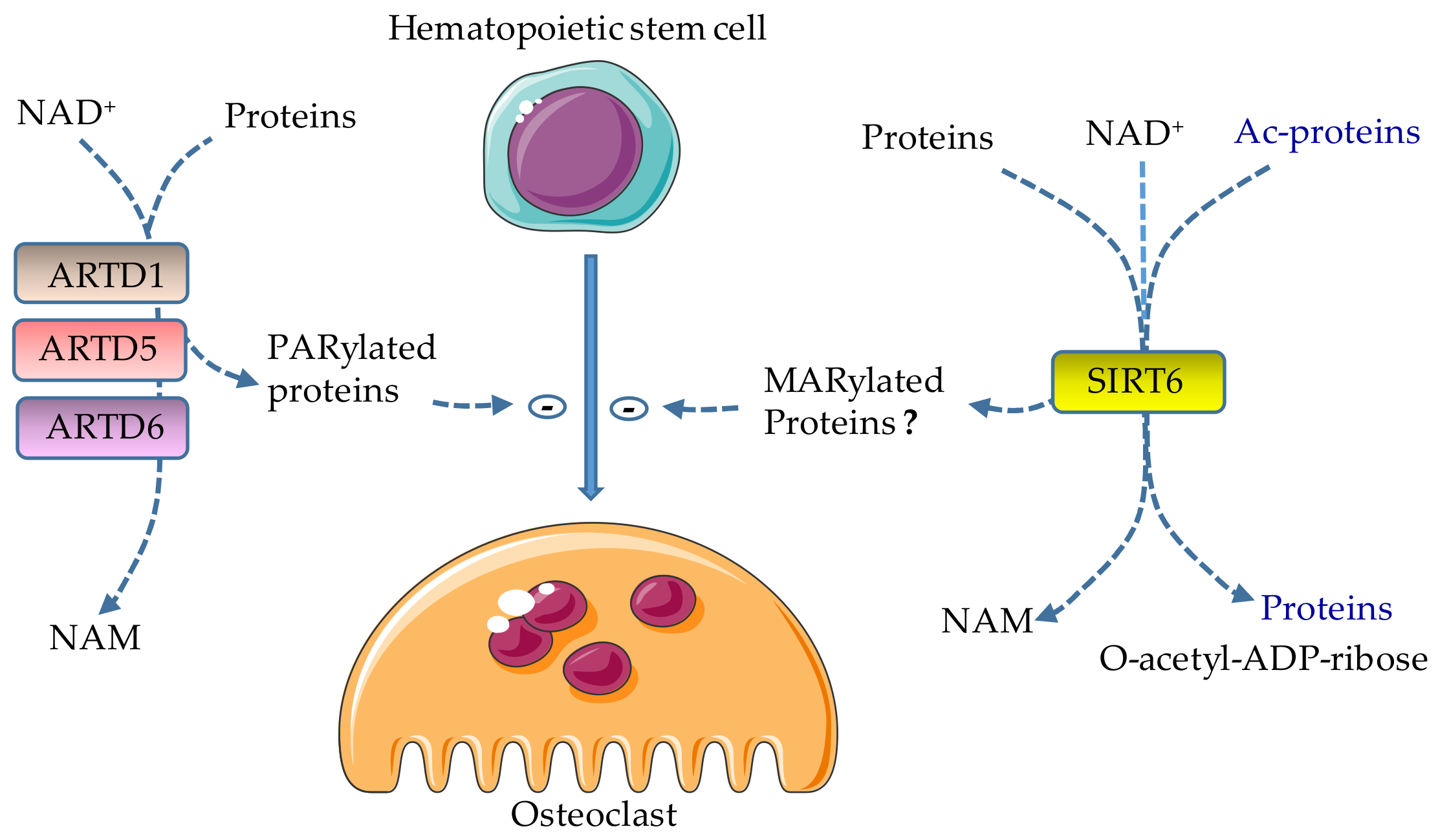
2.2. Role of ARTD5 and ARTD6 in Osteoclast Differentiation
3. Therapeutic Implications
References
- Charbord, P.; Tavian, M.; Humeau, L.; Péault, B. Early ontogeny of the human marrow from long bones: An immunohistochemical study of hematopoiesis and its microenvironment. Blood 1996, 87, 4109–4119.
- Chen, L.T.; Weiss, L. The development of vertebral bone marrow of human fetuses. Blood 1975, 46, 389–408.
- Seike, M.; Omatsu, Y.; Watanabe, H.; Kondoh, G.; Nagasawa, T. Stem cell niche-specific Ebf3 maintains the bone marrow cavity. Genes Dev. 2018, 32, 359–372.
- Alippe, Y.; Wang, C.; Ricci, B.; Xiao, J.; Qu, C.; Zou, W.; Novack, D.V.; Abu-Amer, Y.; Civitelli, R.; Mbalaviele, G. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci. Rep. 2017, 7, 6630.
- Alippe, Y.; Mbalaviele, G. Omnipresence of inflammasome activities in inflammatory bone diseases. Semin. Immunopathol. 2019.
- Charles, J.F.; Hsu, L.-Y.; Niemi, E.C.; Weiss, A.; Aliprantis, A.O.; Nakamura, M.C. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J. Clin. Investig. 2012, 122, 4592–4605.
- Jacome-Galarza, C.E.; Lee, S.-K.; Lorenzo, J.A.; Aguila, H.L. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J. Bone Miner. Res. 2013, 28, 1203–1213.
- Jacquin, C.; Gran, D.E.; Lee, S.K.; Lorenzo, J.A.; Aguila, H.L. Identification of multiple osteoclast precursor populations in murine bone marrow. J. Bone Miner. Res. 2006, 21, 67–77.
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545.
- Gu, R.; Santos, L.L.; Ngo, D.; Fan, H.; Singh, P.P.; Fingerle-Rowson, G.; Bucala, R.; Xu, J.; Quinn, J.M.W.; Morand, E.F. Macrophage migration inhibitory factor is essential for osteoclastogenic mechanisms in vitro and in vivo mouse model of arthritis. Cytokine 2015, 72, 135–145.
- Romas, E.; Bakharevski, O.; Hards, D.K.; Kartsogiannis, V.; Quinn, J.M.W.; Ryan, P.F.J.; Martin, T.J.; Gillespie, M.T. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000, 43, 821.
- Mbalaviele, G.; Veis, D.J. Inflammasomes in Bone Diseases. Exp. Suppl. 2018, 108, 269–279.
- Mbalaviele, G.; Novack, D.V.; Schett, G.; Teitelbaum, S.L. Inflammatory osteolysis: A conspiracy against bone. J. Clin. Investig. 2017, 127, 2030–2039.
- Novack, D.V.; Mbalaviele, G. Osteoclasts-Key Players in Skeletal Health and Disease. Microbiol. Spectr. 2016, 4, 4.
- Kanatani, M.; Sugimoto, T.; Takahashi, Y.; Kaji, H.; Kitazawa, R.; Chihara, K. Estrogen via the Estrogen Receptor Blocks cAMP-Mediated Parathyroid Hormone (PTH)-Stimulated Osteoclast Formation. J. Bone Miner. Res. 1998, 13, 854–862.
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019.
- Lerner, U.H.; Ohlsson, C. The WNT system: Background and its role in bone. J. Intern. Med. 2015, 277, 630–649.
- Sartawi, Z.; Schipani, E.; Ryan, K.B.; Waeber, C. Sphingosine 1-phosphate (S1P) signalling: Role in bone biology and potential therapeutic target for bone repair. Pharmacol. Res. 2017, 125, 232–245.
- Meshcheryakova, A.; Mechtcheriakova, D.; Pietschmann, P. Sphingosine 1-phosphate signaling in bone remodeling: Multifaceted roles and therapeutic potential. Expert Opin. Ther. Tar. 2017, 21, 725–737.
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769.
- Quint, P.; Ruan, M.; Pederson, L.; Kassem, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Sphingosine 1-Phosphate (S1P) Receptors 1 and 2 Coordinately Induce Mesenchymal Cell Migration through S1P Activation of Complementary Kinase Pathways. J. Boil. Chem. 2013, 288, 5398–5406.
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713.
- Sato, K.; Suematsu, A.; Nakashima, T.; Takemoto-Kimura, S.; Aoki, K.; Morishita, Y.; Asahara, H.; Ohya, K.; Yamaguchi, A.; Takai, T.; et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 2006, 12, 1410–1416.
- Huh, J.-E.; Lee, W.I.; Kang, J.W.; Nam, D.; Choi, D.-Y.; Park, D.-S.; Lee, S.H.; Lee, J.-D. Formononetin Attenuates Osteoclastogenesis via Suppressing the RANKL-Induced Activation of NF-κB, c-Fos, and Nuclear Factor of Activated T-Cells Cytoplasmic 1 Signaling Pathway. J. Nat. Prod. 2014, 77, 2423–2431.
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241.
- Yasui, T.; Tsutsumi, S.; Aburatani, H.; Hirose, J.; Nakamura, K.; Tanaka, S. Epigenetic regulation of osteoclast differentiation: Possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J. Bone Miner. Res. 2011, 26, 2665–2671.
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-κB Subunits in the Control of Transcription. Cells 2016, 5.
- Kim, J.H.; Kim, K.; Jin, H.M.; Song, I.; Youn, B.U.; Lee, S.H.; Choi, Y.; Kim, N. Negative feedback control of osteoclast formation through ubiquitin-mediated down-regulation of NFATc1. J. Biol. Chem. 2010, 285, 5224–5231.
- Nayak, A.; Glöckner-Pagel, J.; Vaeth, M.; Schumann, J.E.; Buttmann, M.; Bopp, T.; Schmitt, E.; Serfling, E.; Berberich-Siebelt, F. Sumoylation of the Transcription Factor NFATc1 Leads to Its Subnuclear Relocalization and Interleukin-2 Repression by Histone Deacetylase. J. Boil. Chem. 2009, 284, 10935–10946.
- Chen, N.-M.; Neesse, A.; Dyck, M.L.; Steuber, B.; Koenig, A.O.; Lubeseder-Martellato, C.; Winter, T.; Forster, T.; Bohnenberger, H.; Kitz, J.; et al. Context-Dependent Epigenetic Regulation of Nuclear Factor of Activated T Cells 1 in Pancreatic Plasticity. Gastroenterology 2017, 152, 1507–1520.e15.
- Yasui, T.; Hirose, J.; Aburatani, H.; Tanaka, S. Epigenetic regulation of osteoclast differentiation. Ann. N. Y. Acad. Sci. 2011, 1240, 7–13.
- Beranger, G.E.; Momier, D.; Rochet, N.; Quincey, D.; Guigonis, J.M.; Samson, M.; Carle, G.F.; Scimeca, J.C. RANKL Treatment Releases the Negative Regulation of the Poly(ADP-Ribose) Polymerase-1 on Tcirg1 Gene Expression During Osteoclastogenesis. J. Bone Miner. Res. 2006, 21, 1757–1769.
- Beranger, G.E.; Momier, D.; Guigonis, J.-M.; Samson, M.; Carle, G.F.; Scimeca, J.-C.; Guigonis, J.; Scimeca, J. Differential Binding of Poly(ADP-Ribose) Polymerase-1 and JunD/Fra2 Accounts for RANKL-Induced Tcirg1 Gene Expression During Osteoclastogenesis. J. Bone Miner. Res. 2007, 22, 975–983.
- Beranger, G.E.; Momier, D.; Rochet, N.; Carle, G.F.; Scimeca, J.C. Poly(adp-ribose) polymerase-1 regulates Tracp gene promoter activity during RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 2008, 23, 564–571.
- Chen, J.; Sun, Y.; Mao, X.; Liu, Q.; Wu, H.; Chen, Y. RANKL Up-regulates Brain-type Creatine Kinase via Poly(ADP-ribose) Polymerase-1 during Osteoclastogenesis. J. Boil. Chem. 2010, 285, 36315–36321.
- Petrilli, V.; Herceg, Z.; Hassa, P.O.; Patel, N.S.; Paola, R.D.; Cortes, U.; Dugo, L.; Filipe, H.-M.; Thiemermann, C.; Hottiger, M.O.; et al. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J. Clin. Investig. 2004, 114, 1072–1081.
- Robaszkiewicz, A.; Qu, C.; Wisnik, E.; Ploszaj, T.; Mirsaidi, A.; Kunze, F.A.; Richards, P.J.; Cinelli, P.; Mbalaviele, G.; Hottiger, M.O. ARTD1 regulates osteoclastogenesis and bone homeostasis by dampening NF-kappaB-dependent transcription of IL-1beta. Sci. Rep. 2016, 6, 21131.
- Wang, C.; Xu, C.-X.; Alippe, Y.; Qu, C.; Xiao, J.; Schipani, E.; Civitelli, R.; Abu-Amer, Y.; Mbalaviele, G. Chronic inflammation triggered by the NLRP3 inflammasome in myeloid cells promotes growth plate dysplasia by mesenchymal cells. Sci. Rep. 2017, 7, 4880.
- Wang, C.; Qu, C.; Alippe, Y.; Bonar, S.L.; Civitelli, R.; Abu-Amer, Y.; O Hottiger, M.; Mbalaviele, G. Poly-ADP-ribosylation-mediated degradation of ARTD1 by the NLRP3 inflammasome is a prerequisite for osteoclast maturation. Cell Death Dis. 2016, 7, e2153.
- Oláh, G.; Szczesny, B.; Brunyánszki, A.; López-García, I.A.; Gero, D.; Radak, Z.; Szabo, C. Differentiation-Associated Downregulation of Poly(ADP-Ribose) Polymerase-1 Expression in Myoblasts Serves to Increase Their Resistance to Oxidative Stress. PLoS ONE 2015, 10, e0134227.
- Castri, P.; Lee, Y.J.; Ponzio, T.; Maric, D.; Spatz, M.; Bembry, J.H. Poly(ADP-ribose) polymerase-1 and its cleavage products differentially modulate cellular protection through NF-κB-dependent signaling. BBA-Mol. Cell Res. 2014, 1843, 640–651.
- Zerfaoui, M.; Errami, Y.; Naura, A.S.; Suzuki, Y.; Kim, H.; Ju, J.; Liu, T.; Hans, C.P.; Kim, J.G.; Elmageed, Z.Y.A.; et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 2010, 185, 1894–1902.
- Valdor, R.; Schreiber, V.; Saenz, L.; Martínez, T.; Muñoz-Suano, A.; Domínguez-Villar, M.; Ramírez, P.; Parrilla, P.; Aguado, E.; Garcia-Cozar, F. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871.
- Kameoka, M.; Ota, K.; Tetsuka, T.; Tanaka, Y.; Itaya, A.; Okamoto, T.; Yoshihara, K. Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem. J. 2000, 346, 641–649.
- Olabisi, O.A.; Soto-Nieves, N.; Nieves, E.; Yang, T.T.C.; Yang, X.; Yu, R.Y.L.; Suk, H.Y.; Macian, F.; Chow, C.-W. Regulation of Transcription Factor NFAT by ADP-Ribosylation. Mol. Cell. Boil. 2008, 28, 2860–2871.
- Malireddi, R.K.S.; Ippagunta, S.; Lamkanfi, M.; Kanneganti, T.-D. Cutting edge: Proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol. 2010, 185, 3127–3130.
- Qu, C.; Bonar, S.L.; Hickman-Brecks, C.L.; Abu-Amer, S.; McGeough, M.D.; Peña, C.A.; Broderick, L.; Yang, C.; Kading, J. NLRP3 mediates osteolysis through inflammation-dependent and -independent mechanisms. FASEB. J. 2015, 29, 1269–1279.
- Erener, S.; Pétrilli, V.; Kassner, I.; Minotti, R.; Castillo, R.; Santoro, R.; Hassa, P.O.; Tschopp, J.; Hottiger, M.O. Inflammasome-Activated Caspase 7 Cleaves PARP1 to Enhance the Expression of a Subset of NF-κB Target Genes. Mol. Cell 2012, 46, 200–211.
- García, S.; Bodaño, A.; González, A.; Forteza, J.; Gómez-Reino, J.J.; Conde, C. Partial protection against collagen antibody-induced arthritis in PARP-1 deficient mice. Arthritis Res. Ther. 2006, 8.
- Kunze, F.A.; Bauer, M.; Komuczki, J.; Lanzinger, M.; Gunasekera, K.; Hopp, A.K.; Lehmann, M.; Becher, B.; Müller, A.; Hottiger, M.O. ARTD1 in Myeloid Cells Controls the IL-12/18–IFN-γ Axis in a Model of Sterile Sepsis, Chronic Bacterial Infection, and Cancer. J. Immunol. 2019, 202, 1406–1416.
- Oliver, F.J.; Murcia, J.M.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454.
- Burkart, V.; Wang, Z.-Q.; Radons, J.; Heller, B.; Herceg, Z.; Stingl, L.; Wagner, E.F.; Kolb, H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999, 5, 314–319.
- Mabley, J.G.; Jagtap, P.; Perretti, M.; Getting, S.J.; Salzman, A.L.; Virag, L.; Szabo, E.; Soriano, F.G.; Liaudet, L.; Abdelkarim, G.E.; et al. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm. Res. 2001, 50, 561–569.
- Hottiger, M.O. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu. Rev. Biochem. 2015, 84, 227–263.
- Levaot, N.; Voytyuk, O.; Dimitriou, I.; Sircoulomb, F.; Chandrakumar, A.; Deckert, M.; Krzyzanowski, P.M.; Scotter, A.; Gu, S.; Janmohamed, S.; et al. Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 2011, 147, 1324–1339.
- Ueki, Y.; Lin, C.-Y.; Senoo, M.; Ebihara, T.; Agata, N.; Onji, M.; Saheki, Y.; Kawai, T.; Mukherjee, P.M.; Reichenberger, E.; et al. Increased Myeloid Cell Responses to M-CSF and RANKL Cause Bone Loss and Inflammation in SH3BP2 “Cherubism” Mice. Cell 2007, 128, 71–83.
- Levaot, N.; Simoncic, P.; Dimitriou, I.; Scotter, A.; Rose, J.L.; Willett, T.; Ng, A.; Wang, C.; Janmohamed, S.; Grynpas, M.; et al. 3BP2 deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J. Clin. Invest. 2011, 121, 3244–3257.
- Fujita, S.; Mukai, T.; Mito, T.; Kodama, S.; Nagasu, A.; Kittaka, M.; Sone, T.; Ueki, Y.; Morita, Y. Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone 2018, 106, 156–166.
- Jiang, Q.; Paramasivam, M.; Aressy, B.; Wu, J.; Bellani, M.; Tong, W.; Seidman, M.M.; Greenberg, R.A. MERIT40 cooperates with BRCA2 to resolve DNA interstrand cross-links. Genes Dev. 2015, 29, 1955–1968.
- Nagy, Z.; Kalousi, A.; Furst, A.; Koch, M.; Fischer, B.; Soutoglou, E. Tankyrases Promote Homologous Recombination and Check Point Activation in Response to DSBs. PLoS Genet. 2016, 12, 1005791.
- Zhong, L.L.; Ding, Y.; Bandyopadhyay, G.; Waaler, J.; Börgeson, E.; Smith, S.; Zhang, M.; Phillips, S.A.; Mahooti, S.; Mahata, S.K.; et al. The PARsylation activity of tankyrase in adipose tissue modulates systemic glucose metabolism in mice. Diabetologia 2016, 59, 582–591.
- Yeh, T.-Y.J.; Beiswenger, K.K.; Li, P.; Bolin, K.E.; Lee, R.M.; Tsao, T.-S.; Murphy, A.N.; Hevener, A.L.; Chi, N.-W. Hypermetabolism, Hyperphagia, and Reduced Adiposity in Tankyrase-Deficient Mice. Diabetes 2009, 58, 2476–2485.
- Mariotti, L.; Pollock, K.; Guettler, S. Regulation of Wnt/beta-catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636.
- Guettler, S.; LaRose, J.; Petsalaki, E.; Gish, G.; Scotter, A.; Pawson, T.; Rottapel, R.; Sicheri, F. Structural Basis and Sequence Rules for Substrate Recognition by Tankyrase Explain the Basis for Cherubism Disease. Cell 2011, 147, 1340–1354.
- Ueki, Y.; Tiziani, V.; Santanna, C.; Fukai, N.; Maulik, C.; Garfinkle, J.; Ninomiya, C.; Doamaral, C.; Peters, H.; Habal, M.; et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 2001, 28, 125–126.
- Mukai, T.; Fujita, S.; Morita, Y. Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2. Cells 2019, 8, 195.
- Yoshitaka, T.; Mukai, T.; Kittaka, M.; Alford, L.M.; Masrani, S.; Ishida, S.; Yamaguchi, K.; Yamada, M.; Mizuno, N.; Olsen, B.R.; et al. Enhanced TLR-MYD88 signaling stimulates autoinflammation in SH3BP2 cherubism mice and defines the etiology of cherubism. Cell Rep. 2014, 8, 1752–1766.
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. New Engl. J. Med. 2016, 375, 2154–2164.
- Marchetti, C.; Imperiale, L.; Gasparri, M.L.; Palaia, I.; Pignata, S.; Boni, T.; Bellati, F.; Panici, P.B. Olaparib, PARP1 inhibitor in ovarian cancer. Expert Opin. Investig. Drugs 2012, 21, 1575–1584.
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435.
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010, 376, 235–244.
- Mizutani, A.; Yashiroda, Y.; Muramatsu, Y.; Yoshida, H.; Chikada, T.; Tsumura, T.; Okue, M.; Shirai, F.; Fukami, T.; Yoshida, M.; et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018, 109, 4003–4014.
- Paulsen, J.E.; Pedersen, N.M.; Kries, J.P.V.; Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Eide, T.J.; et al. A Novel Tankyrase Inhibitor Decreases Canonical Wnt Signaling in Colon Carcinoma Cells and Reduces Tumor Growth in Conditional APC Mutant Mice. Cancer Res. 2012, 72, 2822–2832.
- Li, C.; Zheng, X.; Han, Y.; Lv, Y.; Lan, F.; Zhao, J. XAV939 inhibits the proliferation and migration of lung adenocarcinoma A549 cells through the WNT pathway. Oncol. Lett. 2018, 15, 8973–8982.
- Cheng, H.; Li, X.; Wang, C.; Chen, Y.; Li, S.; Tan, J.; Tan, B.; He, Y. Inhibition of tankyrase by a novel small molecule significantly attenuates prostate cancer cell proliferation. Cancer Lett. 2019, 443, 80–90.
- Jia, J.; Qiao, Y.; Pilo, M.G.; Cigliano, A.; Liu, X.; Shao, Z.; Calvisi, D.F.; Chen, X. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the Hippo cascade. PLoS ONE 2017, 12, e0184068.
- Stratford, E.W.; Daffinrud, J.; Munthe, E.; Castro, R.; Waaler, J.; Krauss, S.; Myklebost, O. The tankyrase-specific inhibitor JW74 affects cell cycle progression and induces apoptosis and differentiation in osteosarcoma cell lines. Cancer Med. 2014, 3, 36–46.
- Martins-Neves, S.R.; Paiva-Oliveira, D.I.; Fontes-Ribeiro, C.; Bovée, J.V.; Cleton-Jansen, A.-M.; Gomes, C.F. IWR-1, a tankyrase inhibitor, attenuates Wnt/β-catenin signaling in cancer stem-like cells and inhibits in vivo the growth of a subcutaneous human osteosarcoma xenograft. Cancer Lett. 2018, 414, 1–15.
- Gröschel, S.; Hübschmann, D.; Raimondi, F.; Horak, P.; Warsow, G.; Fröhlich, M.; Klink, B.; Gieldon, L.; Hutter, B.; Kleinhenz, K.; et al. Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat. Commun. 2019, 10, 1635.


